

0. INTRODUCTION
Tu gagneras ton pain à la suer de ton front, dit la Bible, jusqu’à ce que tu retournes dans le sol d’où tu as été tiré, car tu es poussière et tu retourneras à la poussière. Cette phrase simple a une signification si profonde que la plupart de personnes sur cette terre ne la saisisse pas et d’autres font semblant de la comprendre spirituellement en prétextant à tort ou à raison que les choses spirituelles ne sont pas jugées.
Comprenons qu’en effet, Dieu après avoir créé la terre, il la mise à la disposition de l’homme pour que ce dernier l’exploite et en tire profit pendant son bref séjour. Pour que l’homme tire profit de la terre, il faudrait que ce dernier la transforme d’où même la définition de la chimie : elle est science de l’ensemble des principes et des méthodes permettant une transformation profonde de la matière en vue de son utilisation ; or pour transformer une chose, il faut en avoir de connaissance suffisante. C’est ainsi que la chimie étudie avant tout les propriétés physicochimiques de la matière avant d’entamer sa transformation intime. Reconnaissons qu’à des époques bien reculées, les explications seraient des expériences. D’où simplement, la chimie est donc une science qui étudie la matière, ses propriétés physiques et chimiques et sa transformation intime. On sait que le papier provient du bois, le savon du mélange soude caustique et huile, le sucre de la canne à sucre et de la maîtrise des méthodes scientifiques, l’homme semble devenir le maître de la nature ; il sait que le corps humain contient exactement les éléments chimiques, qui sont restitués au sol après la mort. Par exemple, un arbre fruitier sur une tombe se nourrit vraisemblablement de la sève constituée des éléments chimiques qui ont été restitués au sol par l’homme mort, et les autres vivants qui vont consommer ces fruits, ils vont y récupérer les éléments chimiques dont leur corps ont besoin pour la survie, avec un risque de conclure qu’on mange les éléments constitutifs de leur semblable ou simplement qu’on mange nos semblables. Il s’agit d’un cycle admissible on démontre de manière très nette que rien ne se perd et rien se crée, mais tout se transforme, même les selles qui font partie de l’homme sont utilisées comme engrais.
Un élément peut s’associer à un ou plusieurs autres éléments pour donner des corps simples ou complexe et chaque corps peut se présenter sous trois formes d’Etat physique ou de la matière à savoir : l’Etat liquide (l’eau), l’Etat gazeux (l’air) et l’Etat solide (la pierre).
Partout nous faisons la chimie sans le savoir : préparer la nourriture, chauffer l’huile de palme pour changer sa couleur et son goût, les produit pharmaceutique, c’est de la chimie, …
C’est pourquoi la chimie est une science qui s’occupe de la matière (inerte et vivante), de ses propriétés et de ses transformations. C’est donc une science indispensable à l’ingénieur et fait partie des quatre sciences naturelles que sont : la chimie, la physique, la biologie et la géologie.
Les sciences naturelles étudient la Nature et les phénomènes qui s’y déroulent. Elles sont par essence interdisciplinaires et expérimentales.

Les quatre sciences naturelles avec quelques-unes des sciences dérivées.
La chimie s’intéresse particulièrement aux transformations de la matière, comme, par exemple, à la production d’un composé à partir d’éléments ou à la transformation d’une substance en une autre. Une réaction chimique familière est la transformation du clou en fer en rouille et la décomposition de l’eau en hydrogène et oxygène. Un phénomène physique n’est pas lié à la transformation d’un composé (Exemple : L’ébullition de l’eau, la congélation de l’eau en glace)
0.1. Les grands domaines de la chimie
La chimie est une science vaste et on la divise volontiers en plusieurs branches :
ü La chimie organique s’occupe essentiellement de molécules contenant les éléments carbone, hydrogène, oxygène et azote ; la chimie bio-organique étudie les molécules intervenant dans les organismes vivants (peptides, protéines, par exemple).
ü La chimie inorganique (anciennement « minérale ») s’intéressait principalement à la matière inanimée et à tous les éléments de la table périodique ; aujourd’hui, son champ d’étude inclut également les systèmes vivants (chimie bio-inorganique).
ü La biochimie s’intéresse aux processus se déroulant dans les organismes vivants, par exemple dans les cellules.
ü La chimie physique, à laquelle on rattache parfois la chimie théorique, développe des méthodes d’investigation et essaie de comprendre les phénomènes chimiques d’un point de vue théorique (thermodynamique, cinétique, par exemple).
ü La chimie analytique représente une démarche visant à reconnaître les substances ou les éléments tant du point de vue qualitatif que quantitatif. Elle utilise des méthodes physico-chimiques (instrumentales) ou chimiques (organiques et inorganiques).

0.2. Pourquoi Etudier La Chimie
La chimie est l'étude des propriétés des substances et de la manière dont celles-ci réagissent entre elles.
Tout ce qui vous entoure et ce que nous sommes sont en fait les résultats des réactions chimiques ou substances chimiques. Il est donc impératif pour nous d'étudier ce dont nous sommes faits, c'est-à-dire les substances chimiques pour en savoir leurs propriétés et en tirer profit. Ces différentes substances se retrouvent en nous comme c'est dit plus haut et dans notre vie de chaque jour comme par exemple les engrais chimiques, les médicaments, les plastiques, les aliments, les vêtements faits avec les fibres synthétiques, les savons, les détergents, les colles, les vernis, les peintures, matériaux de construction (barres de fer, aciers, tôles, clous, béton, ciment, etc ), explosifs, solvants, armes chimiques…
0.3. La Chimie Est Une Science Expérimentale
La chimie est une science expérimentale qui se fonde sur la méthode scientifique. La méthode scientifique consiste essentiellement à trouver une réponse aux questions scientifiques grâce à des expériences planifiées exécutées.
0.4. La chimie repose sur des mesures quantitatives
Antoine Lavoisier fut le premier chimiste qui comprit l'importance qu'il y avait à effectuer des mesures quantitatives en chimie. Il découvrit la loi de conservation de la masse : lors d'une réaction chimique, la masse totale des substances mises en réaction est égale à la masse totale des produits formés.
La matière qui compose le globe terrestre existe sous la forme de trois états : solide, liquide ou gazeux.
ü
état
solide (sa forme est
indépendante du récipient qui le contient).
Il existe des contacts étroits entre les différentes particules constituantes
du solide, ce qui en diminue la mobilité. Lorsque les particules sont
régulièrement disposées, elles forment un réseau cristallin (exemple : le
chlorure de sodium NaCl). Dans le cas contraire, on obtient un solide amorphe.
La masse spécifique d'un solide est comprise entre 1 et 20 g/cm3.
ü
état
liquide
(il prend la forme du récipient qui le contient).
Il existe des interactions attractives faibles entre les particules qui le
constituent et qui restreignent leurs mobilités. La densité est de l'ordre de 1
g/cm3.
ü
état
gazeux (il
occupe spontanément tout le récipient qui le contient).
Les particules sont indépendantes et entrent de temps en temps en collision. La
densité d’un gaz est donc très faible (10-3 g/cm3). La particularité d’un gaz
est qu’il peut être compressé de façon significative (par exemple, une
bouteille de gaz liquide de type butane ou propane).

Chapitre 1er : Atomistique
1.1. Rappel des unités de base
La plus part de propriété de la matière sont quotidienne, c’est-à-dire elles sont associées à de nombre qui représente une quantité mesurée dans une certaine unité.
Dans le système international d’unité il existe 7 unités de base :
|
GRANDEUR PHYSIQUE |
UNITE |
SYMBOLE |
|
Masse |
Kilogramme |
Kg |
|
Longueur |
Mètre |
M |
|
Temps |
Seconde |
S |
|
Température |
Kelvin |
K |
|
Quantité |
Mole |
Mol |
|
Intensité du courant électrique |
Ampère |
A |
|
Intensité de la lumière |
Candela |
Cd |
Pour ces unités, il existe des multiples et sous multiple qui sont indiqué sous forme des préfixes.
|
Préfixe |
Symbole |
Multiple |
|
exa |
E |
1018 ou 1000000000000000000 |
|
Péta |
P |
1015 ou 1000000000000000 |
|
Téra |
T |
1012 ou 1000000000000 |
|
Giga |
G |
109 ou 1000000000 |
|
Méga |
M |
106 ou 1000000 |
|
Kilo |
K |
103 ou 1000 |
|
Hecto |
H |
102 ou 100 |
|
Déca |
Da |
10 ou 10 |
|
Déci |
D |
10-1 ou 0,1ou 1/10 |
|
Centi |
C |
10-2ou 0,01 ou 1/100 |
|
Milli |
M |
10-3 ou 0,001 ou 1/1000 |
|
micro |
m |
10-6 ou 0,000001 ou 1/1000000 |
|
Nano |
N |
10-9 ou 0,000000001 ou 1/1000000000 |
|
Pico |
P |
10-12 ou 0,0000000000001 ou 1/1000000000000 |
|
femto |
F |
10-15 ou 0,000000000000001ou 1/1000000000000000 |
|
Atto |
A |
10-18 ou 0,000000000000000001 ou 1/1000000000000000000 |
Les unités dérivés et anciennes
|
GRANDEUR |
SYMBOLE |
UNITE |
DEFINITION |
|
Energie Volume Pression Pression |
E V P P |
Joule Litre Pascal Atmosphère ou bar |
Newtonx mètre 1dm3 = 10-3 m3 = 1l Nm-2 1Atm = 1,018 1bor = 10-5posc |
Les Constantes Fondamentales
Nombre d’Avogadro = 6,023.1023 particules
Unité de masse atomique 1uma = 1,6605872 10-24 gr
1gr = 6,02214199.1023 uma
Constante de Planck h = 6,62607876.10-34 J.s
Vitesse de lumière c = 2,99792456.108 ms-1
= 310 ms-1
Constante de gaz parfait R = 0,8258 Latm.mol-1
= 8,314472 J.mol-1 K-1
Masse de l’électron (ē) = 9,10938188.10-28
= 5,485799.10-4 Uma
Masse du proton = 1,00729 Uma
= 1,67262158.10-24 gr
Masse du Neutron = 1,008649 Uma
= 1,6492716.10-24 gr
1.2. Définitions
La matière : c’est toute chose qui a une masse et qui occupe un volume dans l’espace.
Exemple : une pierre,…
Une matière est constituée des substances ; la substance est composée des molécules, les molécules des atomes et l’atome des électrons et du noyau, le noyau des protons et des neutrons.
Actuellement il existe 118 atomes différents qu’on appelle élément chimique.
1.3. Structure de L’atome
1.1.1 Définition
Le mot atome vient du grec « Atomos » qui veut dire indivisible. Bien que la molécule reste jusqu’aujourd’hui la plus petite particule de la matière qui puisse garder la propriété de cette matière, on s’est aperçu que ces briques élémentaires des molécules, étaient eux-mêmes constitués de particules plus petites, électrons et noyau (lui-même formé de protons et neutrons).
Une question à laquelle il est encore très difficile de répondre est de savoir comment se sont formés les atomes et les molécules. Depuis quelques temps, les physiciens sont convaincus que l’univers a commencé par une grande explosion originelle, le « big-bang ».
1.1.2 Constitution de l’Atome
Les expériences faites par les scientifiques admettent que l’atome est indivisible mais peut être décomposé en particule beaucoup plus petite encore : le noyau et les électrons.
Donc, le noyau très dense, chargé positivement, entouré d’électrons de charge électrique négative. Le noyau est constitué de deux types de particules, les protons et les neutrons appelées nucléons.
La masse des électrons est 1837 fois plus faible que celle des nucléons. Ainsi la masse de l’atome est concentrée dans le noyau et est environ égale à la somme masses des nucléons.



a) Le noyau
Découvert en 1910 par Ernest RUTHERFORD, il est plus petit que l’atome avec une taille qui dia métrique de 210-4 A0, avec une charge positive qui augmente avec la masse atomique. Il comprend le proton et le neutron.
a.1. Le proton
Particule chargé positivement avec une valeur de +1,602.10-19 C et sa masse vaut 1,6726485.10-27 Kg = 1,007276470Uma. C’est lui qui donne sa charge au noyau.
a.2. Le neutron
Est la particule élémentaire sans charge, comme le dit le nom, sa charge est nul (0) et sa masse vaut 1,6749543.10-27 Kg = 1,008665012Uma.
b) Les électrons
Découvert en 1897 par le physicien Allemand JJ. THOMSON, l’électron est défini comme la charge unitaire des ions en solution, avec la charge des particules du rayonnement catholique.
Il est chargé négativement soit -1,60210-19C avec une masse de 9,109534.10-31 = 5,4858.10-4 Uma.
1.1.3 Caractéristique de l’Atome
Par définition un atome est un ensemble électriquement neutre dans lequel on distingue un noyau positif et l’électron électrique négatif, il se caractérise par :
v Son symbole : qui représente chaque élément chimique dont l’une en lettre majuscule et la seconde en minuscule s’il en existe. Les lettres proviennent généralement du nom de l’élément.
Exemple :
- Hydrogène : H – Carbone : C
- Hélium : He – Oxygène : O
- Lithium : Li – Chlore : Cl
v Son numéro atomique ou nombre atomique : c’est son numéro d’ordre dans le tableau périodique, et correspond au nombre total d’électron associé au noyau atomique et au nombre des protons à l’intérieur du noyau, il est symbolisé par Z.
v Sa masse atomique (Ma) : c’est un nombre abstrait, nombre presque décimal car il s’agit d’un rapport de l’isotope 12 du carbone, en l’arrondissant il représente le nombre de masse A. La masse atomique résulte d’une moyenne des masses isotopique relative (mi).
![]()
Où xi est l’abondance isotopique des différents isotopes dans un élément naturelle (en %).
v Le royau atomique : en le considérant comme une sphère sa valeur est d’ordre de 1 à 3 A°. Et se calcule par la relation suivante :
![]()
Avec : n : couche électronique, Ԑ : permittivité, m : masse, h : constante de Planck, e= charge de l’électron.
En conclusion pour avoir la masse de l’atome on peut utiliser la formule suivante :
![]()
Z = Nombre atomique
N = Nombre des neutrons.
A= La masse atomique
Cela représente la sommes des masse des protons et neutrons pas ceux d’électrons car elles sont négligeables.
Notons qu’on parle de Nucléide si l’atome à le même nombre de proton et de neutron, il se représente de la manière suivante :
![]() Ou X est le symbole de
l’élément
Ou X est le symbole de
l’élément
![]()
Il existe des nuclides :
- D’isotope : si l’atome a le nombre de protons et neutron différent, d’où le A est aussi et seulement vous variant.
Exemple : ![]()
Donc ![]()
- D’isobare : si les atomes ont le même nombre de masse A mais ayant un Z différent et correspond à des éléments différents.
Exemple : ![]()
Donc ![]()
-
D’isotone : ici nous avons ![]()
Exemple : ![]()
-
D’isomères : si ![]()
Il appartient au même nuclide, l’un est à l’état dit fondamentale et l’autre à l’état exister par rapport au fondamentale au caractérisé par un état dit « Métastable »
Exemple : ![]()
1.1.4 Hypothèse concernant l’édifice atomique
Le chercheur Anglais JJ THOMSON émis en conséquence, en 1898, la première hypothèse concernant l’édifice atomique, il assimile chaque atome à une minuscule boule compacte chargé positivement et dans laquelle les électrons seraient en fouis comme les grains dans la figure.

1.1.5 Modèle atomique de RUTHERFORD : Modèle planétaire
Pour vérifier le modèle proposé par JJ THOMSON, RUTHERFORD conçut l’idée de bonborder une mince feuille d’Aluminium, d’une épaisseur d’environ un certaine d’atomes, avec les particules alpho (x), émise par le potassium. Pour obtenir un faisceau bien délité de royaux x, Rutherford interposa entre la source de rayonnement et la feuille de métal une épaisse plaque de plomb, munie d’une ouverture circulaire.


Rutherford avait d’abord trouvé que les rayons α subissaient une légère dispersion (déviation) lorsqu’il traversait une mince couche de métal. Des recherches plus poussées feront apparaître que quelques particules (1/104) α subissaient une déviation beaucoup grande. La quasi-totalité des rayons α traversa la feuille métallique sans déviation et une minuscule partie de ces rayons subits un recul. Il conclut que les charges devaient se trouver dans les atomes métalliques sous forme très condensée.
Sur base de ces faits, Rutherford édifie un nouveau modèle atomique totalement différent de celui de Thomson où presque toute la masse de l’atome est concentré dans une partie centrale extrêmement petite et dans laquelle s’accumulent les charges positives. Cette partie centrale s’appelle le noyau atomique et autour de ce dernier gravitent les électrons dont les charges négatives neutralisent les charges positives : « modèle planétaire »
En conclusion
1) L'atome est essentiellement constitué de vide: d' où le fort taux de passage des particules alpha à travers la feuille d'or.
2) L'atome, électriquement neutre, est constitué d'une partie chargée positivement, qui est très petite: c'est ce qui explique la répulsion forte, quoique rare au plan statistique, des particules alpha, de même nature électrique que le "noyau". Ainsi donc, il y a dans l'atome un "noyau", positif, tout petit, autour du quel gravite un nuage négatif, constitué d'électrons. C’est-à-dire qu’il ressemble au système solaire

3) En tournant autour du noyon, l’électron émet continuellement de la lumière parce que d’après les lois classiques de l’électromagnétique (théorie de Max Well), une charge électrique en mouvement émet un rayonnement électromagnétique. Ceci constitue une erreur de la part de Rutherford car l’électron en émettant de l’énergie et tomberait sur le nayon.

1.4. STRUCTURE ELECTRONIQUE DE L’ATOME
1.4.0. Introduction
Les écrits d' Albert EINSTEIN (né en 1879 à Ulm, Allemagne, mort en 1955 à Princeton, Etats-Unis) datent de 1905, l' année de toutes les révolutions: première révolution russe en janvier 1905, fusillade du "9" (calendrier "julien") ou "22" (calendrier "grégorien"), janvier 1905 à Saint-Pétersbourg, mutinerie du cuirassé POTEMKINE en Mer Noire, séparation de l' Eglise et de l' Etat en France.....
Il n'avait alors que... 26 ans. Le talent n'attend pas forcément le nombre des années...
La mécanique dite "classique", datant de GALILEE, de NEWTON, se trouvait incapable de décrire correctement les objets se déplaçant à une vitesse proche de celle de la lumière, dont EINSTEIN démontre qu'elle ne peut être dépassée.
Masse et énergie sont reliées par une formule condensée qui a fait le tour du
monde:
E = m.c2
La masse m d'un objet n'est plus un invariant. Elle est reliée à la masse m 0, au repos, par la relation:
m = ![]()
où v désigne la vitesse de l'objet.
EINSTEIN a reçu le Prix NOBEL de Physique en 1921 pour ses travaux.
EINSTEIN a quitté l'Allemagne en 1933, au moment de la prise du
Pouvoir par les nazis.
Durant la Seconde Guerre mondiale il prit une part active dans le
programme entamé par ROOSEVELT de construction de la bombe atomique, dont la
première tomba sur Hiroshima le 6 août 1945.
Après la Seconde Guerre mondiale, au moment de la guerre froide, il
a largement contribué à défendre la Paix et à montrer que les savants ne sont
pas forcément responsables des utilisations qui sont faites par d'autres de
leurs découvertes: bref, sur un même registre, faut-il en vouloir aux
Chinois d'avoir inventé la poudre alors que cette dernière servait pour faire,
à l' origine, des.... feux d'artifice?
1.4.1
Phénomène lumineux
L'effet photoélectrique est très utilisé dans les systèmes d'alarme qui équipent bon nombre d'établissements en contact avec le public. Le passage d'une personne là "où il ne faut pas" déclenche toutes les alarmes....
Là aussi, cette expérience, son explication, ont provoqué une véritable révolution: jusqu' alors la lumière avait été décrite comme un phénomène de nature "ondulatoire": durant tout le XIX° siècle, de FRESNEL à YOUNG, la lumière était considérée comme un système d'ondes.
Il apparaît alors que la lumière est aussi "corpusculaire", c'est à dire composée de "grains".
C'est cet aspect corpusculaire qui a permis de comprendre ultérieurement l'effet COMPTON, par exemple.
On parle alors pour la lumière d'aspect "dual", de "dualité onde-corpuscule": deux choses en une même chose.
Pour l' heure les physiciens n'ont pas réussi à expliquer par une seule même théorie ce qu'est la lumière. Selon les besoins on privilégie l'aspect "ondulatoire" de la lumière, et selon d'autres moments, on privilégie plutôt l'aspect "corpusculaire".
Mais Einstein revient et dit que la lumière peut être considérer comme étant soit de nature ondulatoire ou soit corpusculaire comme l’a dit ses prédécesseur.
A. Nature ondulatoire
La lumière peut être considérée comme étant constitué d’une vibration électromagnétique qui se propage en onde.
Si dans le vide, l’onde à une vitesse C, elle part par seconde une distance équivalente à un certain nombre des fréquences.
 |
![]()
Avec C : Célérité
![]() : Longueur d’onde
: Longueur d’onde ![]()
![]() : Fréquence
: Fréquence
Les différentes espèces de radiation se distinguent par la
valeur de leur longueur d’onde, la fréquence est le nombre d’onde ![]() , donc :
, donc : ![]()
σ: Nombre d’Onde
B. Nature corpusculaire
Cet effet a été découvert par le physicien allemand HERTZ (cfr les ondes "hertziennes") vers 1885.
On irradie par une lumière monochromatique, de fréquence n0 croissante, une plaque métallique reliée à un électromètre à cadran, déchargé à l' instant initial.
L'électromètre commence à se charger au-delà d'une fréquence n0. Les deux branches de l'électromètre s'écartent, signe de la présence d'électricité de même nature dans les plaques.
Que s'est-il passé?
Lorsqu' on éclaire une plaque métallique et qu'on procède à un balayage en fréquence pour la lumière on obtient une émission d'électrons à partir d'une fréquence-seuil n0 qui est caractéristique du métal utilisé. C'est EINSTEIN qui a apporté cette explication à l'expérience d’HERTZ. Il s'était écoulé environ 20 ans entre les deux événements....
D' après EINSTEIN la lumière est porteuse de grains de matière, les "quanta", appelés aussi "photons", porteurs chacun d' une énergie E qui est égale au produit de deux termes: la constante de PLANCK et la fréquence de la radiation.
E = h.n
La constante de PLANCK vaut 6.62.10-34 J.s et n, la fréquence de la radiation, est exprimée en hertz, symbole Hz, dans le système SI.
Ces grains d'énergie viennent, selon EINSTEIN, cogner contre les atomes métalliques de la plaque de l'électromètre et, s'ils ont suffisamment d'énergie, arrachent des électrons de la plaque, d'où la production d'électricité.
C'est ce qui constitue l'effet "photoélectrique", c'est à dire la production, grâce à la lumière, d'électricité.
Si l'on éclaire une plaque métallique avec une lumière monochromatique de fréquence n supérieure à la fréquence-seuil n0, le surcroît d'énergie par rapport au travail d'extraction W0, tel que:
W0 = h.n0
est dissipé sous forme d'énergie cinétique prise par les électrons:
E apportée par les photons monochromatiques = h.n = h.n0 + 0.5.m.v2
1.4.2 Spectres atomique
On appelle spectre, un ensemble des raies (rayonnement) résultant de la décomposition d’une lumière.
On distingue 2 sortes de spectre :
a) Spectre continue
Lorsqu’on fait passer de la lumière blanchâtre à travers un prisme on observe la décomposition de la lumière en divers faisceau de couleur différente. On obtient ainsi un spectre continu ou chaque couleur se fond (confusion) avec ses voisins (cas de l’Arc-en-ciel)
b) Spectre discontinu
b.1 spectre d’émission
Lorsque des gaz ou vapeur d’une substance sort portés à haute température, il se produit une émission de lumière. La résolution de la lumière émise par un prisme conduit à un spectre d’émission. Ce spectre est discontinu et est composé d’un ensemble des voies distincte et donc de fréquence différente.

b.2 Spectre d’absorption
Si on place un corps quelconque sur le trajet d’un faisceau lumineux issu d’une source dont le spectre est continu, la réalisation de cette lumière par un prisme donné à spectre de voies correspondant exactement au spectre d’émission. C’est le spectre d’absorption.
C. FORMULE EMPIRIQUES
Le spectre atomique d’hydrogène fut particulièrement étudié par Johann BALMER (physicien Suisse 1885). Il trouva que les longueurs d’onde des voies peuvent être calculées par la relation suivante :
![]()
RH : constante de Rudberg pour l’atome d’Hydrogène ;
n : nombre entier supérieur à 2.
En général, RUDBERG proposa la formule suivante :
![]()
RH : 13,6 eV = 21,8.10-19
Avec une série de voies pour :
n1 = 1 série de Lyman
n1 = 2 série de Balmer
n1 = 3 série de Pashen
n1 = 4 série de Brackett
n1 = 5 série de Pfund


1.4.3 Le modèle atomique selon Niels BOHR
L'idée de Niels BOHR (physicien danois né en 1887), et qui ne fut pas contestée au début, était la suivante: l'atome, avec son noyau et ses électrons, doit tout siumplement être un "système solaire miniature": le noyau figurant le Soleil et les électrons les planètes qui gravitent autour.
1.
Expression
du rayon Rn de l'atome d'hydrogène, dans la théorie de
BOHR.
L'électron
est soumis, dans ce modèle, à deux forces extérieures: son poids Pet
la force de COULOMB, F.
Le poids P est négligeable devant la force coulombienne.
La force de COULOMB a pour expression:
F = 
Où e0 est la permittivité du vide: 8.85.10-12 F.m-1.
BOHR postule alors que les trajectoires de l'électron sont circulaires.
A partir de là, en calquant sur l'atome d'hydrogène les lois de la mécanique
classique, il écrit:
La somme vectorielle des forces extérieures appliquées à l'électron est égale
au produit de sa masse par l'accélération du mouvement.
Soit:
SExtérieures = m.a
Avec:
a = a tangentielle + anormale
Or,
dans le cas d'un mouvement circulaire uniforme, ce qui est postulé ici,
l'accélération tangentielle est nulle. L’accélération a se
confond alors avec l’accélération normale.
Avec alors : ![]()
A
partir de là on écrit:![]()
A partir de là BOHR postule que le moment cinétique sde l’électron est un multiple entier de la constante de PLANCK réduite:
s = m.v.R = ![]() (2)
(2)
L'égalité écrite en (2) constitue un trait de génie. BOHR, en effet, fait un lien alors entre l'Ancien (le moment cinétique exprimé dans le cadre de la mécanique classique) et le Moderne (la quantification des propriétés de la matière).
A partir de là, le reste n'est plus que "tripatouillage" de données.
On va s'arranger pour faire disparaître v et on isolera R qu'on exprimera en fonction des données.
m.v2 =
![]() (3)
(3)
D'
où: v2 = ![]() (4)
(4)
Or,
d' après l'équation (2):
v =![]() (5)
(5)
On
élève (5) au carré et on obtient:
v2 = ![]() (6)
(6)
En
identifiant alors (4) et (6) on arrive, en extrayant R, à: R = ![]() (7)
(7)
On remplace numériquement e0, h, e par leurs valeurs respectives, et on arrive à:
Rn= 5.3.10-11.n2 m (8)
Soit:
R n = 0.53.n2 ![]() (9)
(9)
On constate que le rayon des orbites de l'atome d'hydrogène est "quantifié". Il ne peut prendre que des valeurs discrètes, comme par exemple:
R1 =
0.53.12![]()
R2 = 0.53.22 ![]() = 2.12
= 2.12![]()
Les valeurs autres que celles données par la formule (9) ne sont pas possibles.
On appelle n le "nombre quantique principal". C'est un entier naturel, non nul.
2. Expression de l'énergie En de l'atome d'hydrogène.
Un des acquis fondamentaux de la mécanique classique a été d'énoncer le fait que l'énergie totale E d'un système mécanique est égale à la somme de deux termes, l'énergie cinétique Ec et l'énergie potentielle Ep.
E = Ec + Ep
On a : Ec = 0.5.m.v2
Or,
d' après l'équation (3), m.v2 est égal à: ![]()
D' où, bien entendu: Ec = 0.5.m.v2 = 0.5.![]() (10)
(10)
Que se passe-t-il au niveau de l'énergie potentielle Ep?
Tout
se passe comme si un opérateur imaginaire venait tirer sur l'électron, en
orbite autour du noyau, dans la même direction que la force coulombienne, MAIS
dans un sens opposé. Cet opérateur imaginaire amènerait alors l'électron de la
distance R à la distance plus l'infini.
Le travail W effectué par la force exercée par cet opérateur imaginaire serait
donné par l'intégrale suivante:
W =![]() (11)
(11)
Par
définition, l' énergie potentielle Ep du système "électron-noyau" de
l' atome d' hydrogène est égale à l' opposé de l' intégrale W.
L' énergie potentielle Ep est l' énergie que met en oeuvre l'
électron pour résister à l' arrachement de celui-ci par l' opérateur imaginaire
qui voudrait le porter de la distance R à la distance plus l' infini.
C'est donc l' énergie qu' il a en lui, son énergie "potentielle", qui
lui permet de résister.
Ep =
- ![]() (12)
(12)
Tous calculs faits, en posant que l'énergie potentielle est nulle lorsque l'électron est à la distance plus l'infini, on trouve:
Ep = ![]() (13)
(13)
On remarque que l'énergie potentielle du système est négative: cela tient au choix de l'origine de Ep, nulle lorsque R est à l' infini.
En réunissant les résultats de (10) et (13) on arrive à l'énergie totale du système:
E totale = ![]() (14)
(14)
On
peut alors remplacer R par son expression trouvée en (7):
On arrive, tous calculs et réarrangements faits, à:
E totale =
![]() (15)
(15)
En remplaçant chacun des termes par sa valeur numérique on arrive alors à une énergie totale, mesurée en joules, pour l'atome d'hydrogène, qui est égale à:
E totale =
![]() J (16)
J (16)
Si l'on pose qu'un électronvolt est égal à 1.6.10-19 J, (16) s'écrit sous la forme usuelle que tout le monde retient:
E totale =
![]() eV (17)
eV (17)
3. Synthèse
En appliquant les lois de la physique classique, le modèle de Rutherford était erronés, car l’électron devait tomber sur le noyon, or les atomes sont stable et présentent un spectre discontinu.
En 1913, BOHR reprend le modèle de RUTHERFORD pour en donner une explication raisonnable de la stabilité et de l’existence des spectrales et écorce l’incompatibilité apparente, il introduit des conditions qui excluent l’application des lois de physique classique au phénomène atomique. Ces axiomes dont connu sous le nom des conditions de BOHR.
Elles sont les suivantes :
1° L’énergie de l’électron sur orbite est négative c’est-à-dire que l’électron ne peut pas quitter de soi-même l’atome. Donc pour lui séparer de l’atome il faut lui donner une autre énergie.
2° L’énergie de l’électron sur une orbite permise est constante, elle ne varie seulement que lorsque l’électron saute d’une orbite à l’autre.
3° La variation d’énergie de l’électron correspond à l’émission ou à l’absorption d’un quantum d’énergie.

4° Il y a émission de la lumière (énergie) lors que l’électron saute d’une orbite d’énergie supérieure à une orbite d’énergie inférieure (n2 → n2).
5° Il y a absorption d’énergie lors que l’électron quitte l’orbite d’énergie inferieure à l’orbite d’énergie supérieure (n2 → n2).
6° Dans les conditions normales c’est-à-dire à l’état stable (atome non excité), l’électron se place toujours sur l’orbite n = 1 pour le cas de l’hydrogène (r = 0,53) et des hydrogènoïdes (H+, li+2).
7° L’électron passe de l’orbite n2 d’énergie E2 à l’orbite n1 d’énergie E1.
![]()
![]()
Avec ![]() = fréquence, h =
constante de PLANCK
= fréquence, h =
constante de PLANCK
Or ![]()
1.4.4 Insuffisances de la Théorie de Bohr et Sommer Feld
La structure planétaire de l’atome proposée par Rutherford s’est révélée insuffisante pour interpréter et expliquer les spectres atomiques.
En outre, le spectre de l’atome serait continu, l’expérience nous montre que le spectre est toujours discontinu.
La théorie de Bohr explique les spectres de l’atome d’hydrogène qui ne comporte qu’un seul électron, quand l’atome d’hydrogène est excité dans un champ magnétique ou électrique, on observe des déplacements, ou même des nouvelles voies non prévisibles par la théorie de Bohr. Sommer Feld interprète ce nouveau phénomène en définissant pour chaque valeur n, un ensemble d’orbite elliptique, il introduit ainsi pour repérer l’état énergétique de l’électron dans l’atome, des nombreux quantiques supplémentaire, l et m.
Notons que la théorie de Bohr même compléter par celle de Sommer, ne parvient pas à interpréter les spectres des atomes lourd. Ce modèle est presque dépassé mais permet de retrouver par calculs simples certaines relations utiles.
1.4.5 Les nombres quantiques : introduction à la mécanique quantique
Pour décrire le comportement de l’électron et préciser son énergie, on a généralement besoin de quatre nombre quantiques.
A. Nombre quantique principal « n »
C’est le numéro d’ordre de différentes orbites permises sur lesquelles gravitent les électrons. L’énergie de l’électron dépend uniquement de ce nombre.
Au lieu d’être représenté par des chiffres (1,2,3,…), les niveaux sont caractérisés par des lettres représentants les couches :
n = 1 couche K
n = 2 couche L
n = 3 couche M
n = 4 couche N
n = 5 couche O
n = 6 couche P
n = 7 couche Q
Il indique l’énergie ou la taille de l’orbite.
B. Le nombre quantique secondaire « l »
![]() Pour un même niveau
d’énergie principale, il existe des sous niveaux d’énergie qu’on appelle des
couches qui sont représenté par les lettres : s, p, d, f. Leur nombre par
niveau est donné par la relation suivante :
Pour un même niveau
d’énergie principale, il existe des sous niveaux d’énergie qu’on appelle des
couches qui sont représenté par les lettres : s, p, d, f. Leur nombre par
niveau est donné par la relation suivante :
Exemple :
K : n = 1, l = 0 sous couche s
L : n = 2, l = 0 sous couche s
l = 1 sous couche p
M : n = 3, l = 0 sous couche s
l = 1 sous couche p
l = 2, sous couche d
N : n = 4, l = 0 sous couche s
l = 1 sous couche p
l = 2 sous couche d
l = 3 sous couche f
Notons qu’une orbite est un volume de l’espace où l’on a une très grande probabilité d’avoir ou de trouver un électron. Ce nombre quantique secondaire détermine la forme de l’orbite ou orbitale.
C. Le nombre quantique magnétique « m »
Etudié par ZEEMAN, dès 1896. Lorsqu’un spectre est tiré dans un champ magnétique, d’autre voies apparaissent, celle-ci, montrent l’orientation des différentes sous couches dans l’espace, c’est nombre magnétique « m » en varie de – l à +l et leur nombre est de 2l + 1.

D. Nombre quantique de spin « s »
Emise par GOUDSMIT et UHZENBECK en 1985, lors que l’électron tourne autour du noyau, il tourne aussi autour de lui-même et :
- Il peut tourner soit dans le sens de sa révolution autour du noyau ;
- Soit dans le sens inverse de celui de sa révolution autour du noyau ;
Ces deux mouvements ont des énergies différentes. Même si la différence est très faible, il existe deux nombre quantiques spin.
![]()
Représentation Spatiale Des Sous Couches
1° Sous couche s : il en existe une seul sorte d’orbitale s parce qu’une sphère ne peut être représenter que d’une seul manière par rapport aux axe x,y et z.

2° Sous couche p : ils sont symétrique par rapport aux axe x, y, z, donc il en existe 3 types

3° Sous couche d : elles sont symétriques par rapport au plan (x,y) (y,z), donc il en existe 5 sortes selon cette orientation
²
4° Les orbites f ont des formes très complexes qu’on ne peut pas représenter dans l’espace à 3 dimensions.
1.4.6 Remplissage Des Couches Et Sous Couches Electronique
A. Principe d’exclusion de PAULI
Un atome et toujours constitué de Z proton et électron et N neutre. Chacun des électrons est caractérisé par 4 nombres quantiques qui lui sont propre. Le principe de PAULI énoncé en 1925 régie la représentation des électrons sur les différentes couches : « Il est impossible de trouver dans un même atome 2 électrons caractérisé par 4 nombre quantique identique, ils doivent pour de moins différer par la valeur de l’un de ces nombres »
B. Règle de HUND
Elle est stipulé de la manière suivante : « Le remplissage des cases quantiques ou orbital d’une sous couche en formation s’effectue de telle sorte qu’on ait le maximum des spins parallèle dans cette sous couche »
Par convention on représente la structure électronique d’un atome dans son état fondamental par 2 modèles :
1°) On le fait en écrivant les 2 premiers nombres quantique n,sx ou x est le nombre d’électrons de la sous couche.
Exemple : Z = 7 2s2 2s2 2p3
2°) On le fait par des cases dites case quantiques
 .
.
C. Déviation des niveaux d’énergie : règle de KLECHKO WSKY
Elle permet de retrouver facilement l’ordre dans lequel se remplissent les niveaux d’énergie électronique à l’état fondamental c’est-à-dire l’état correspond à l’énergie minimale de l’atome ou état stable de l’atome.

![]()

Donc le nombre maximum d’électron par sous couche est :
ü Pour la sous couche s=2
ü Pour la sous couche p=6
ü Pour la sous couche d=10
ü Pour la sous couche f=14
1.5. CLASSIFICATION PERIODIQUE DES ELEMENTS
La classification périodique des éléments a apporté à la chimie la 1ère base d’une construction rationnelle et elle est pour le chimiste une véritable charte et une référence pérennante. Elle est née de l’observation expérimentale, bien avant que la structure électronique des atomes ne sois connue, à partir du constant qu’il existe des analogies entre les caractères chimiques de certains corps simples.
Premièrement proposé par le Russe DIMITRI IVANOVITCH MEDELEÏV en 1869 avec 63 éléments qui étaient déjà connu à l’époque, il les a classés en tenant compte de leur masse atomique croissante. Après la connaissance de la structure atomique, cela à inciter à représenter l’idée de classification des éléments basés sur le numéro atomique, c’est-à-dire sur le nombre des électrons tournant autour du noyau.
15.1 La 1ère période : H et He
L’Hydrogène possède un seul électron dans l’orbite 1s et l’Hélium en a deux, par conséquent c’est la couche K.
15.2 La 2ème période : Li à Ne
Elle commence avec le Lithium qui remplit totalement l’orbital 1s et entame 2s, ce dernier étant séparé du noyau par une couche saturée et ne ressent plus l’attraction du noyau et cet électron est plus facilement arrachable pour donner Li+ ce qui caractérise les alcalins, et il en est de même pour le Bore qui commence à remplir progressivement l’orbital 2p jusqu’au Néon un autre gaz rare pour qui la couche L devient saturée.
15.3 La 3ème période :
Avec le Sodium (2 = 11) commence le remplissage de la couche M jusqu’à l’Argon (2 =18) un autre gaz inerte pour lequel la couche M est complètement saturée comme ses homologues précédent.
15.4 La 4ème période : K à Kr
Elle s’ouvre avec le Potassium (2 =19), puis vient un élément alcalinaux-terreux, le calcium. En effet, après le Co vient le Sc (Z = 2) : la sous couche 3d commence donc à être rempli et l’on a 8 éléments dit de transitions jusqu’au Ni (2 = 28). Les éléments de transitions contiennent toute les sous 3d incomplètes. Ce phénomène leur confère des propriétés paramagnétique et ferromagnétique (Fr, Co, Ni) intéressante. En suit viennent le Cu et le Zn remplissant la sous couche 3d, leur configuration fait d’eux un pseudo-alcalin et un pseudoalcanino-terreux. La sous couche 4s étant très voisine en énergie par rapport à la sous couche 3d avec le Cr (2 = 24) et Cu (Z = 29), et enfin le Ga reprend le remplissage de la sous couche 4p jusqu’au Kr un autre gaz inerte.
15.5 La 5ème période : Rb à Xe
Partant du Rubidium on trouve une période de 18 éléments contenant 8 autres éléments de transition de l’Ittrium au Palladium, remplissant progressivement la sous couche 4d, le 2 éléments suivants l’Argent et le Cadnium (Cd) ressemble à leur homologue Cu et Zn avec l’In commence le remplissage de la couche 5p qui se termine avec le Xe un autre gaz inerte.
15.6 La 6ème période : Cs à Rm
Elle est très longue avec 32 éléments. En effet, elle commence avec Cs et le Ba, remplissage de l’orbital 4f demeurée vide jusqu’à présent, avec une série de 14 autres éléments de propriétés voisines : les LANTHANIDES du Cérium au Cutecium, puis viennent les éléments de transition, ceux de la famille du Platine suivie d’un pseudo-alcalin l’Or (Au) et d’un pseudo-alcalino-terreux le Hg. En fin la période se termine par un remplissage progressif de la sous couche 6p du Thallium jusqu’au Radom dont la sous couche est saturée.
14.7 La 7ème période : à partir de Fr
Il commence le remplissage de l’orbitale 5f. Ces éléments ont ses propriétés voisines à d’autre mis appart les éléments qu’on appelle les ACTINIDES dont une partie sont des éléments artificiel, le TRANSURANIENS.

Chapitre 2ème : Les Liaisons Chimiques
2.1. Introduction et Définition
On appelle liaison chimique toute interaction attractive qui maintient des atomes à courte distance. Cette interaction peut être directionnelle comme la liaison entre deux atomes au sein d'une molécule, ou non-directionnelle comme l'interaction électrostatique qui maintient les ions d'un cristal ionique au contact. Elle peut être forte comme les deux précédents exemples, ou faible comme les interactions de van der Waals qui sont de nature dipolaire.
De nombreux modèles existent pour décrire ces interactions. Par exemple la liaison chimique entre deux atomes au sein d'une molécule peut être décrite avec le modèle de Lewis ou avec un modèle quantique, comme la théorie des orbitales moléculaires. Dans les deux cas, l'origine de l'interaction est un partage d'électrons entre les deux atomes partenaires de la liaison chimique.
Les liaisons plus faibles sont expliquées, en général, par des polarités entre des molécules. C'est le cas des interactions très faibles comme les forces de London qui font partie des forces de van der Waals. De telles forces interprètent le maintien dans un état condensé solide ou liquide de composés moléculaires comme le diiode ou les hydrocarbures.
La description d'une liaison chimique doit préciser le modèle utilisé et l'énergie de la liaison.
Les liaisons
Décrire pourquoi les atomes des molécules ou des cristaux demeurent au contact est l'objet de l'étude de la liaison chimique. Si les liaisons chimiques n'existaient pas (ou quand elles ne sont pas assez solides par rapport à l'énergie de température), les atomes ne restent pas au contact. C'est l'état liquide, voire gazeux. Comprendre ce qu'est une liaison chimique permet d'interpréter la réaction chimique. En effet, une réaction chimique n'est autre que la transformation des liaisons chimiques.
Il existe un grand nombre de façons de décrire les liaisons chimiques. D'une part, chaque type de liaison fait appel à un modèle différent :
- Modèle pour la liaison covalente ;
- Modèle pour la liaison métallique ;
- Modèle pour la liaison hydrogène ;
etc.
D'autre part, pour un type de liaison donnée, il existe plusieurs modèles :
- Pour la liaison covalente :
- Pour la liaison entre un ligand et un ion métallique dans un complexe :
- Modèle du champ cristallin
- Modèle de champ des ligands
- Sans parler des modèles qui ne sont plus utilisés comme le modèle des liaisons datives et d'autres.
etc.
L'intérêt d'avoir plusieurs modèles pour un même type de liaison relève souvent de l'histoire des sciences. Par exemple, le modèle de Lewis est venu très tôt (1916) alors que le modèle des orbitales moléculaires (par exemple LCAO-MO, ce qui signifie Linear Combinaison of Atomic Orbitale [pour faire des] Molecular Orbitals) a été introduit plus récemment. Il est bien plus complexe à utiliser mais il fournit de nombreuses informations sur la liaison covalente que ne fournit pas le modèle de Lewis, par exemple il permet de prévoir, par le calcul, l'énergie de la liaison ou la prévision du spectre électronique. Quand de telles informations ne sont pas utiles, le modèle de Lewis suffit et est utilisé bien que moins performant et plus ancien.
Le développement théorique le plus abouti, utilisé pour décrire une liaison chimique, est la théorie des orbitales moléculaires. Celle-ci décrit les atomes par des fonctions appelées orbitales atomiques. Des combinaisons linéaires de ces fonctions constituent les orbitales moléculaires qui décrivent les molécules. Ces orbitales moléculaires peuvent être :
- liantes ; dans ce cas, les électrons de liaison ont la plus grande probabilité de se trouver entre les noyaux qu'ailleurs ; l'orbitale tend alors à maintenir les noyaux ensemble ; ces orbitales sont l'équivalent des liaisons covalentes de la théorie de Lewis ;
- non-liantes : dans ce cas, les électrons ont plus de probabilité de se trouver plus près d'un des noyaux ; ces orbitales sont l'équivalent des doublets non liants de la théorie de Lewis.
- antiliantes : dans ce cas les électrons ont plus de probabilité de ne pas se trouver entre deux atomes liés ; ces orbitales n'ont pas d'équivalent dans la théorie de Lewis.
La liaison ionique s'interprète différemment. Elle se rencontre dans un cristal et maintient les anions (négatifs) et les cations (positifs) au contact. Des forces électrostatiques maintiennent les ions de signes opposés au contact alors que des forces de même nature entre les ions de même signe tendent à faire "éclater" le cristal. Il se trouve que la somme des forces attractives est plus grande que la somme des forces répulsives ; le cristal peut ainsi exister.
Pour les modèles qui donnent accès à l'énergie des atomes et des molécules, les liaisons se forment (et donc les édifices qu'elles constituent existent) si l'énergie de l'édifice (molécule, cristal) est inférieure à l'énergie des atomes ou des ions pris séparément. Ainsi, l'énergie de la molécule H2 est inférieure à l'énergie de deux atomes H. La molécule H2 existe donc. En revanche, l'énergie de la molécule He2 est supérieure à l'énergie de deux atomes d'hélium pris séparément. Ceci explique que la molécule He2 n'existe pas.
2.2. Calcul de l'énergie de la liaison de la molécule de dihydrogène
Le premier calcul de l'énergie d'une liaison chimique, fondateur de la Chimie quantique est celui de la molécule la plus simple, celle de l'hydrogène par Bohr en 1913. C'est sans doute le seul calcul de liaison chimique accessible à un non-spécialiste.

Il consiste à appliquer le
modèle de Bohr de l'atome à une molécule. On fait l'hypothèse que les électrons
ont un mouvement circulaire de rayon ![]() autour de l'axe des protons p+p+, supposés immobiles et distants de
R. La distance électron-proton e-p+ est
autour de l'axe des protons p+p+, supposés immobiles et distants de
R. La distance électron-proton e-p+ est ![]() . En utilisant la formule du modèle de Bohr de l'atome pour l’état
fondamental :
. En utilisant la formule du modèle de Bohr de l'atome pour l’état
fondamental :
![]()
Où p=mv est la quantité de mouvement et
![]()
La constante de Planck réduite, l'énergie cinétique des électrons s'écrit :

Le
potentiel V est attractif entre électrons et protons et se compose des quatre
liaisons électron-proton. Il y a répulsion entre les électrons distants de ![]() et les
protons distants de R. L'énergie potentielle s'écrit:
et les
protons distants de R. L'énergie potentielle s'écrit:

L'énergie totale est:

Dans un atome d’hydrogène, l’égalité entre la force électrostatique et la force centrifuge peut s’écrire :
![]()
où ![]() est
la constante de Rydberg,
est
la constante de Rydberg,
![]() Å le
rayon de Bohr de l'atome d'hydrogène
Å le
rayon de Bohr de l'atome d'hydrogène
et ![]() la
constante diélectrique.
la
constante diélectrique.
En y retranchant l’énergie de liaison ![]() de deux atomes d’hydrogène isolés, l’énergie totale de la molécule
devient :
de deux atomes d’hydrogène isolés, l’énergie totale de la molécule
devient :

où ![]() et
et ![]() .
.
Cette équation se résout graphiquement en faisant varier x de telle façon que l'énergie du minimum soit minimale. On obtient ainsi x = 1,15 et y = 2,7 ce qui donne les valeurs trouvées par Bohr en 1913 de 2,7 eV pour l’énergie de liaison et de 0,6 Å pour l'écartement des protons. La précision du calcul est certes médiocre puisque les valeurs expérimentales sont, respectivement de 4,5 eV et de 0,74 Å. On trouvera des méthodes plus perfectionnées basées sur les orbitales moléculaires.
2.3. L’électronégativité
Le concept électronégativité a été introduit par PAULING en 1931. C’est une mesure de la capacité d’un atome dans une molécule à attirer un électron à lui-même. Il s’agit d’un nombre sous dimension calculée à partir des forces des liaisons entre molécule. Elles indiquent ainsi la capacité pour deux atomes de partager leur électron de valence. Notons ainsi qu’un élément qui perd facilement un ou plusieurs électrons est dit électropositif et celui qui en gagne est électronégatif
Ou encore l'électronégativité d'un élément désigne la force d'attraction que cet élément exerce sur les électrons d'une liaison. L'élément le plus électronégatif est le Fluor (4) et le moins électronégatif est le Césium (0,7) et donc le Césium est plus électropositif que le Fluor. L'électronégativité des autres éléments varie entre ces deux mesures. L'électronégativité augmente de gauche à droite dans une période et de bas en haut dans une même famille. L'électronégativité par le symbole c.
Alors l’élément le plus électronégatif du tableau périodique est le Fluor et le moins électronégatif est le Francium en d’autre mot le Francium est l’élément du tableau périodique le plus électropositif et le Fluor le moins électropositif.
|
|
|
|
|||||||||||||||
|
|
|
|
|
|
|
|
|
|
|||||||||
|
|
|
|
|
|
|
|
|
||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||

2.4. La liaison Electrovalente ou l’électrovalence ou encore liaison ionique
L’électrovalence est le transfert d’électron d’un atome électropositif vers un atome électronégatif ou le transfert d’électron des atomes métalliques vers les atomes non métalliques en vue de réaliser l’octet. En général les atomes des familles Ia, IIa et IIIa donnent les électrons aux atomes des familles VIa et VIIa
Ex : ![]()

La liaison électrovalente est une hétéro polaire c’est-à-dire la liaison entre particule de signe opposé. La molécule ionique est toujours polaire ou dipolaire. La valence ionique est la charge portée par un ion, elle correspond au nombre d’électron porté par un ion, elle correspond aussi au nombre d’électron perdu ou capté ; elle est positive si l’atome perdu un électron et négative si l’atome en a gagner. Ce qui fait qu’un ion positif est plus petit que son atome et l’ion négatif est grand que son atome.
2.5. La liaison covalente ou atomique
Une liaison covalente est une liaison chimique dans laquelle deux atomes partagent deux électrons (un électron chacun ou deux électrons venant du même atome) d'une de leurs couches externes afin de former un doublet d'électrons liant les deux atomes. C'est une des forces qui produit l'attraction mutuelle entre atomes.
La liaison covalente implique généralement le partage équitable d'une seule paire d'électrons, appelé doublet liant. Chaque atome fournissant un électron, la paire d'électrons est délocalisée entre les deux atomes. Le partage de deux ou trois paires d'électrons s'appelle respectivement « liaison double » et « liaison triple ».
Une liaison covalente est une liaison dans laquelle deux électrons de valence sont partagés entre deux non-métaux. Dans ce type de liaison, il doit y avoir une différence d’électronégativité inférieure à 1,7 sur l’échelle de Pauling. Il y a formation d’une liaison covalente (sauf pour les liaisons de coordinence), lorsqu’il y a un recouvrement de deux orbitales atomiques ayant chacune un électron de valence. Grâce à ce recouvrement, il y aura la formation d’une seule et unique orbitale. Ce recouvrement d’orbitales atomiques conduit à la formation d’une orbitale moléculaire
La liaison covalente peut-être normale ou dative :
a. Liaison covalente normale
Ici il y a mis en commun de deux électrons, elle peut être
a.1. Homopolaire ou apolaire
Elle se passe entre 2 atomes identiques, comme par exemple l’hydrogène ou l’oxygène :

a.2. Hétéro polaire ou polaire
Elle se passe entre deux atomes différents, en grande majorité ceux ayant une différence d’électronégativité pas très grande mais très proche.

b. Liaison dative
Les donneurs d’électron sont généralement les non métaux ayant atteint une configuration stable d’un gaz inerte par formation d’ion ou par réalisation de plusieurs covalence normale. Les accepteur sont généralement les protons, les sulfurides, les métaux de transition à l’état d’atome où à l’état d’ion, qui compléteront ainsi leur orbitale incomplètes ; cas des complexe.
La covalence Dative peut être :
ü Soit dative coordinative : LCDC : pour ce cas le donneur des doublais est plus électronégatif que l’accepteur. C’est-à-dire le double versait à la communauté revient au donneur après séparation, cette liaison est non polarisé, et donc l’étage d’oxydation du donneur ne change pas.
ü Soit dative semi-polaire : LCDSP : pour ce cas le donneur de doublais électronique est moins électronégative que l’accepteur, ce qui veut dire que l’accepteur est plus électronégatif et attire les doublais vers lui, d’où une apparition des charges partielle sur l’atome ; cette liaison est dite polarisé et par conséquent l’atome donneur des doublais augmente d’étage d’oxydation.
Notons que le nombre de covalence dative est le nombre de doublait électronique libre susceptible d’être donné où accepté par un atome.

2.6. Liaison hydrogène

Ex : Liaison hydrogène entre des molécules d'eau.
La liaison hydrogène ou pont hydrogène est une force intermoléculaire impliquant un atome d'hydrogène et un atome électronégatif comme l'oxygène, l'azote et le fluor L'intensité d'une liaison hydrogène est intermédiaire entre celle d'une liaison covalente et celle des forces de van der Waals.
On pensait à l'origine que l'électron de l'atome d'hydrogène était partagé entre les molécules liées, et donc que cette liaison hydrogène était quasi-covalente. On sait aujourd'hui qu'elle est à 90 % électrostatique. Pour que cette liaison s'établisse, il faut être en présence d'un donneur de liaison hydrogène et d'un accepteur :
- le donneur possède une case quantique vide est composé d'un composé à H acide, c'est-à-dire un atome d'hydrogène lié à un hétéroatome (comme dans les amines, alcools, thiols) ;
- l'accepteur est composé d'un hétéroatome (uniquement azote, oxygène ou fluor) porteur d'un doublet non liant.
Lorsqu'une liaison hydrogène s'établit, les deux hétéroatomes se trouvent à une distance d'environ 0,25 nm.
Conséquences de la liaison hydrogène
La liaison hydrogène s'établit alors entre toutes les molécules présentant les caractéristiques précédemment évoquées ; voyons par exemple le cas d'un acide carboxylique (R-COOH).

Ex : Liaison hydrogène entre des molécules d'acides.
On remarque que toutes les molécules sont liées entre elles au niveau de la fonction alcool. Le radical alkyl « R » aura alors une influence non négligeable sur la force de cette liaison. En effet, la longueur de la chaîne et sa composition vont polariser de façon plus ou moins marquée la liaison entre l'hydrogène et l'oxygène. Si la liaison H (comme on l'appelle plus couramment) est affaiblie, la cohésion inter-moléculaire le sera également et la température d'ébullition de la substance en question sera plus faible. Autrement dit, il faudra moins d'énergie (par le biais de la chaleur) pour séparer les molécules les unes des autres. Au contraire, pour l'eau (H2O), l'ammoniaque (NH3) ou le fluorure d'hydrogène (HF), la liaison X-H est tellement polarisée que les liaisons H qui s'établissent confèrent aux substances des points d'ébullition anormalement hauts.
Une autre illustration peut être celle de l'eau solide (glace). En effet, la molécule d'eau est l'exemple typique de la liaison H. Les liaisons H s'établissent, de sorte que l'état liquide de l'eau est l'état le plus compact, tandis que pour tout autre corps pur c'est l'état solide. Dans la glace, l'eau est en structure tétraédrique (structure rendue possible par ces liaisons), et la compression d'un bloc de glace conduit au retour à l'état liquide. C'est pourquoi la glace occupe plus de volume que l'eau, en quantités égales (le glaçon flotte sur l'eau).

Ex : Liaisons hydrogène entre les molécules polymériques du Kevlar.
Enfin - et bien que la liste ne puisse être exhaustive tant le domaine d'application de cette liaison est vaste - on mentionnera le cas des polymères, tel le Poly-para-phénylène téréphtalamide (plus connu sous le nom de Kevlar). Les chaînes de polymères s'attachent entre elles par des liaisons H lui conférant ainsi ses propriétés si intéressantes de résistance.
En chimie, une force de van der Waals, interaction de van der Waals ou liaison de van der Waals est une interaction électrique de faible intensité entre atomes, molécules, ou entre une molécule et un cristal. Bien qu'il soit possible de décrire sommairement cette interaction en considérant les forces électriques qui sont présentes entre tous les couples de charges électriques qui forment ces atomes et ces molécules en définitive, c'est un phénomène qui ne peut bien se comprendre que dans le cadre de la physique quantique. Ces forces ont été nommées en l'honneur du physicien néerlandais Johannes Diderik van der Waals (1837 — 1923), prix Nobel de physique 1910, qui fut le premier à introduire leurs effets dans les équations d'état des gaz en 1873. On retrouve les effets de cette force à l'extrémité des pattes du gecko, assurant ainsi leur forte adhésion sur du verre.
Les forces de van der Waals ont trois origines :
- L'interaction électrostatique attractive ou répulsive entre deux multipôles permanents selon leurs orientations (effets d'orientation). On les appelle les forces de Keesom.
- L'interaction attractive entre un multipôle permanent et un multipôle induit (effets d'induction). On les appelle les forces de Debye.
- L'interaction électrostatique attractive entre deux multipôles induits (effets de dispersion). On les appelle les forces de London.
L'énergie des forces de van der
Waals ![]() peut donc se formuler de la façon
suivante :
peut donc se formuler de la façon
suivante :
![E_{\text{van der Waals}}=- \frac{1}{r^6} \left [ \underbrace{\frac{\mu_1^2 \cdot \mu_2^2}{3 ( 4 \pi \cdot \epsilon_0 \cdot \epsilon )^2 \cdot k_B \cdot T}}_{E_{\text{Keesom}}} + \underbrace{\frac{\mu_1^2 \cdot \alpha_2 + \mu_2^2 \cdot \alpha_1}{( 4 \pi \cdot \epsilon_0 \cdot \epsilon )^2}}_{E_{\text{Debye}}} + \underbrace{\frac{3}{4} \cdot { \frac{h \cdot \nu \cdot \alpha_1 \cdot \alpha_2}{( 4 \cdot \pi \cdot \epsilon_0)^2} }}_{E_{\text{London}}} \right ]](CHIMIE%20GENERALE%20UAC%202017_fichiers/image100.png)
Les trois termes de cette expression peuvent être décomposés de la manière suivante :
Cette énergie est liée aux forces de Keesom, dues à l'interaction entre deux molécules polaires.
L’interaction dipôle-dipôle est beaucoup plus faible qu’une interaction ion-dipôle puisque l'interaction se produit entre charges partielles. L’énergie potentielle typique de ce type d’interaction est de l’ordre de 2 kJ/mol. Elle varie de façon inversement proportionnelle avec la distance à la puissance 6 entre le centre des dipôles de chacune de ces molécules.
Cette énergie est liée aux forces de Debye, dues à l'interaction entre une molécule polaire et un dipôle induit.
Cette énergie est liée aux forces de London, dues à l'interaction entre deux dipôles induits.
Ici, on a utilisé les notations suivantes :
 est la constante
diélectrique du vide ;
est la constante
diélectrique du vide ; la constante de Planck ;
la constante de Planck ; la constante de Boltzmann ;
la constante de Boltzmann ; la température absolue ;
la température absolue ; la distance moyenne
entre les molécules considérées ;
la distance moyenne
entre les molécules considérées ; les moments dipolaires des molécules considérées ;
les moments dipolaires des molécules considérées ; , la fréquence
électronique d'absorption (Hz) ;
, la fréquence
électronique d'absorption (Hz) ; les polarisabilités
électroniques.
les polarisabilités
électroniques.
Ces forces peuvent s'exprimer de manière différente, lorsque les distances entre les molécules mises en jeu deviennent plus grandes que quelques nanomètres. Il faut alors prendre en compte les effets de retard dus à la propagation de la lumière avec une vitesse finie (forces de Casimir-Polder).
b. L’interaction ion-dipôle et l’hydratation des cations en solutions
L’interaction ion-dipôle résulte de l’attraction d’un anion orientée par la charge partielle positive d’un dipôle ou de l’attraction d’un cation orientée par la charge partielle négative d’un dipôle. L’énergie potentielle est négative et augmente à l’inverse du carré de la distance comme l’indique la formule suivante :
où :
- est le moment
dipolaire ;
 est la charge de
l’ion ;
est la charge de
l’ion ;- est la
permittivité du vide ;
- est la distance
séparant le milieu du dipôle et l’ion.
L’énergie potentielle typique de ce type d’interactions est d’environ 15 kJ/mol. Cette attraction devient nulle à grande distance (entre le dipôle et l’ion). Cette interaction est responsable de l’hydratation des cations en solution3.
Les forces de van der Waals
s'obtiennent en divisant l'expression de l'énergie de van der Waals par ![]() . Il s'ensuit
qu'elles varient en
. Il s'ensuit
qu'elles varient en ![]() .
.
Elles permettent également d'expliquer la rondeur de la Terre ou d'un liquide en apesanteur.
Dans le cas de molécules polaires, cette force s'ajoute à la force purement électrostatique (de même comportement) entre les dipôles permanents. Dans le cas de molécules à symétrie sphérique, d'atomes, etc. la force de van der Waals est la seule qui entre en jeu pour ces distances.
À très
longue distance, où il ne peut plus être question de liaison chimique, les
forces de van der Waals entrent dans le cadre de l'électrodynamique quantique : à courte et longue distance,
elles se décrivent proprement comme dues à l'échange des particules
virtuelles entre les atomes. On entre alors dans le cadre des forces
de Casimir, décroissant en ![]() .
.
Les liaisons de van der Waals n'entrent pas dans le cadre des liaisons chimiques, en ce sens que les électrons restent sur leurs atomes (ou molécules) respectifs (les termes d'échanges restent négligeables). Elles sont l'origine du terme de pression négative intervenant en correctif dans l'équation du gaz parfait. Elles sont essentielles pour appréhender les forces entre atomes de gaz noble.
Pour les très courtes distances on entre alors dans le domaine de la chimie, où les diverses liaisons (liaison hydrogène, liaison métallique...) deviennent compétitives, et peuvent l'emporter.
Les forces de van der Waals participent ainsi à la physisorption, et entrent en jeu dans le phénomène de capillarité.
Les échanges de particules virtuelles, qui sont leur fondement théorique, se retrouvent dans des phénomènes de même type : forces entre deux surfaces.
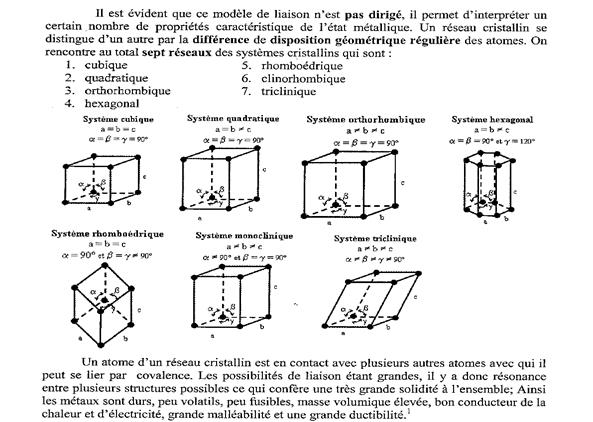
Le maintien en contact des atomes au sein d'un métal s'interprète suivant plusieurs modèles.
- Le modèle le plus simple considère que le métal est constitué non pas d'atomes M, mais de cations M+ ayant mis en commun chacun un électron. C'est cette mise en commun, avec une délocalisation sur tout le cristal métallique, qui rend compte de la stabilité du cristal.
- La théorie des bandes utilise la théorie plus élaborée des orbitales atomiques. On peut comprendre cette théorie en considérant :
- dans un premier temps la molécule Li2 décrite par le recouvrement des orbitales atomiques 2s de chaque lithium. Il se forme ainsi une OM liante et une OM antiliante. Chaque électron 2s se retrouve dans l'OM liante, d'où une stabilité de l'édifice.
- dans un second temps, on considère la molécule Li3 où 3 OA 2s sont mises en commun. Il se forme 3 OM, avec les mêmes conclusions que pour Li2.
- enfin, avec la molécule Lin, n très grand, il se forme n/2 OM liantes et n/2 OM antiliantes. Ces groupes d'OM étant en très grand nombre dans un espace énergétique limité, elles forment une bande d'OM liantes et une bande d'OM antiliantes.
Cette théorie explique non seulement la stabilité du cristal métallique, mais également le pouvoir conducteur électrique du métal, les électrons se déplaçant dans les bandes délocalisés sur tout le cristal.
Chapitre 3ème : Notion de Chimie Nucléaire
3.0. Définition
La chimie nucléaire est une partie de la chimie qui s’occupe des phénomènes nucléaire (du noyau). En d’autre mot la chimie nucléaire s’occupe de la structure de noyau, leur transformation en d’autre noyau et les problèmes de leur synthèse.
C’est ausssi la chimie des éléments radioactifs (éléments chimiques dont tous les isotopes sont radioactifs) tels que les actinides, le technétium, le radium ou le radon, cette chimie étant associée à des équipements spéciaux (tel que les réacteurs nucléaires ou autres…) conçus pour exécuter des processus nucléaires. Cette science inclut l’étude de la corrosion des surfaces et du comportement de matériaux ou d’atomes dans des conditions de fonctionnement normales et anormales (par exemple lors d’un accident). Un autre domaine important de la chimie nucléaire est le comportement des objets et des matériaux radioactifs qui ont été placés dans un site de d’élimination ou de stockage de déchets nucléaires.
Cette chimie inclut l’étude des effets chimiques résultant de l’absorption du rayonnement par les animaux vivant, les plantes et les autres matériaux. La chimie nucléaire a grandement facilité la compréhension des traitements médicaux spéciaux (comme la radiothérapie sur les cancers), et a permis à ces traitements de s’améliorer.
Elle comprend également l’étude de la production et de l’utilisation des sources radioactives et/ou nucléaire dans un éventail de processus liés à la chimie. On peut citer parmi ces processus étudiés la radiothérapie pour des applications médicales, l’utilisation de traceurs radioactifs au sein de l’industrie, des sciences ou encore de l’environnement, la production de combustibles nucléaires ou encore la spectroscopie à résonance magnétique nucléaire (RMN) qui est couramment utilisée en chimie organique de synthèse, en physico-chimie et pour l’analyse structurelle de chimie macromoléculaire... L’utilisation de ces rayonnements est également étudiée pour la modification de matériaux, principalement les polymères. Enfin, elle traite de l’influence de la masse atomique des éléments sur les réactions chimiques et les propriétés des composés chimiques.
3.1. Caractéristiques d’un noyau atomique
On rappelle que la charge élémentaire est e = 1,6.10 - 19 C. En physique nucléaire, on utilise souvent l'unité de masse atomique 1 u = 1, 66. 10 - 27 kg.
La classification des noyaux est premièrement par le nombre atomique, Z. Dans un atome neutre il y a Z électrons, donc Z proton. Z détermine les propriétés chimiques des atomes. On parle donc de noyau d’hydrogène, de noyau de carbone, etc.
Mais les propriétés nucléaires (stabilité avant tout) d’un noyau sont déterminées aussi par le nombre des neutrons qu’il contient. Un nuclide est une espèce de noyau avec un nombre fixe de Z protons et N neutrons. Puisque le proton et le neutron ont à peu près la même masse, la masse d’un nuclide est approximativement donnée pas le nombre de masse, A, où A = Z+N. Les protons et les neutrons ensemble sont appelés les nucléons.
![]()
3.2.Classification des nucléides
Les nucléides sont une catégorie d'atomes définie par le même nombre de masse A, le même numéro atomique Z et le même état énergétique nucléaire. Nous dénombrons environ 325 nucléides naturels (dont 274 sont stables et 51 radioactifs) et environ 1 200 nucléides artificiels. Certains nucléides peuvent être d'origine soit naturelle, soit artificielle : par exemple le soufre 34 (élément stable).
Les nucléides sont classés en fonction soit de leur structure en isotopes, isobares, isotones ou isodiaphères, soit de leurs niveaux énergétiques (même structure) en isomères.
a) Isotopes
Ce sont des atomes dont les noyaux ont un même nombre de protons (Z constant). Par exemples : L’hydrogène comporte 4 isotopes : - l'hydrogène normal : 1H dont le noyau est le proton (p), - l'hydrogène lourd ou deutérium : 2H ou D dont le noyau est le deuton (d), - l'hydrogène très lourd ou tritium : 3H ou T dont le noyau est le triton qui est un isotope radioactif ou radioisotope et l’hydrogène éphémère 4H qui apparaît lorsque on bombarde des noyaux d'hélium 4He avec des photons d'énergie 1 GeV.
le carbone comprend 6 isotopes dont :deux sont stables : 12C et 13C - et quatre sont radioactifs : 10C, 11C, 14C (celui servant à la datation archéologique) et 15C
b) Isobares
Ce sont des atomes dont les noyaux ont un même nombre de masse A. Ainsi, nous avons : le carbone 14 (radioactif) : 14C et l'azote 14 (stable) : 14N
c) Isotones
Nous désignons par isotones des atomes qui possèdent un nombre de neutrons N identique. Par exemple : le carbone 14 (radioactif) : 14C et l'azote 15 (stable) : 15N
d) Isodiaphères
Ce sont des atomes ayant à la fois leurs nombres de neutrons N, de masse A et de protons Z différents, mais dont la différence entre le nombre de neutrons N et le nombre de protons (ou numéro atomique) Z est constante. Par exemple, nous trouvons les groupes suivants : Le carbone 12 (stable) : 12C, l'azote 14 (stable) : 14N et l'oxygène 16 (stable) : 16O
e) Isomères
Nous désignons par isomères des atomes ayant même nombre de masse A, même numéro atomique Z et même nombre de neutrons N mais dont les énergies internes de liaison de leurs nucléons sont différentes. Cette forme instable (donc radioactive) peut être de longue durée et correspond à un état excité qui disparaît par émission d'un photon X ou γ. Exemples : Le technétium 99 : 99Tc radioélément artificiel présente une forme métastable (utilisée en médecine nucléaire) : 99Tcm
3.3. Les différents modes de radioactivité (ou de désintégration)
Au-delà du noyau du bismuth 20783Bi (Z > 83) on ne trouve aucun nucléïde stable. Tout noyau instable va tendre à devenir stable par désintégration ou émission de rayonnement ou de la radioactivité. Toute désintégration radioactive donne naissance à un élément chimique différent.
En fonction de la nature du rayonnement émis, on peut distinguer :
![]() le
rayonnement alpha (α)
le
rayonnement alpha (α)
![]() le
rayonnement bêta - (β–)
le
rayonnement bêta - (β–)
![]() le
rayonnement bêta + (β+)
le
rayonnement bêta + (β+)
![]() le
rayonnement gamma (γ)
le
rayonnement gamma (γ)
a) La désintégration bêta moins (β- )
C’est celle que subissent les noyaux ayant un excès de neutrons, par exemple : 131I par rapport à 127I stable ; en conséquence, il y a alors transformation d'un neutron en un proton et un électron ou "beta moins, β- (n → p + + e-). Le noyau se transforme donc de la manière suivante:
![]()
C’est une transformation isobarique puisque le nombre de masse A est inchangé ;
b) La désintégration bêta plus (β+)
C’est celle que
subissent les noyaux ayant un excès de protons, par exemple: 124I).
Il s'agit de la
transformation d'un proton en un neutron et un positon, β+
(ou anti-électron 0+1e), p → n+ e+. Le noyau se
transforme donc de la manière suivante:
![]()
C’est aussi une transformation isobarique.
c)L'émission alpha
Seuls les noyaux lourds peuvent émettre des particules α parce que ce phénomène nécessite une grande énergie de liaison. Par exemple: 21084Po ; il s'agit de l'émission de 2 protons et de 2 neutrons qui correspond au noyau d'hélium ou particule α.

Ce n'est pas une transformation isobarique.
Exemple 1 : Prédire le type de la décroissance radioactive des isotopes radioactifs suivants:
![]()
Réponse: (a) Ca -40 est l’isotope stable, Ca-47 a un Z+N egal a 47 qui est supérieur a 40 c’est ainsi que le Ca-47 decroit par émission β-.( b) Al-27 est l’isotope stable tandis que Al-25 a un Z+N = 25 qui est inferieur a 27 donc l’isotope Al-25 se désintègre soit par émission d’un positron β+ ; (c) le Na-23 étant l’unique isotope stable de sodium , le Na-26 décroit par émission β- et (d)le Na-23 étant l’unique isotope stable de sodium , Na-22 se desintègre soit par émission d’un positron.
3.4.Les réactions nucléaires
Une réaction nucléaire survient quand une particule de projectile frappe un noyau de cible. La première réaction nucléaire, qui fut aussi la dernière contribution séminale de Rutherford à la physique, était la première transmutation nucléaire
![]()
Deux autres réactions très connues avec des particules a sont la découverte du neutron (Chadwick, 1932),
![]()
et la découverte de la radioactivité artificielle et la désintégration β+(Curie-Jolliot, 1934),
![]()



On écrit l’équation de la réaction nucléaire :
![]()
avec X qui est le noyau cible, a le projectile, Y est le noyau produit et b la ou les particules éjectées (on dit émergeante), à noter que a ou b peuvent également être des photons.
Dans la notation abrégée de Bethe, on écrit le noyau cible, à gauche, entre parenthèses on écrit le projectile et la particule émergente, séparés par une virgule, et finalement le noyau produit. A X (a, b) A’ Y
Une réaction nucléaire est décrite par l’équation symbolique:
![]()
Pour équilibrer une réaction nucléaire on doit simplement appliquer le principe de conservation de masse et en particulier de s'assurer que le nombre de nucléons (protons et neutrons) est conservé.
![]()
23892U (12C, 4n) 24698Cf (californium), 238U (18O,5n) 251100Fm (fermium)
24896 Cm (18O, 5n) 261104Rf (rutherfordium). L’élément Z= 111 a été synthétisé à Darmstadt (17/12/ 1994) par la réaction 20983Bi (64Ni, n) 272111. Le 112 a été synthétisé le 9 février 1996 par la réaction 20882Pb (7030Zn, n) 277112 . Les Z = 114 et 116 ont été découverts en 2000.
Exemple : Compléter les réactions nucléaires suivantes:

Réponse:

3.5.La Radioactivité
La radioactivité est la propriété naturelle de certains noyaux atomiques à émettre de façon spontanée un rayonnement, impliquant la désintégration de noyaux atomiques instables. Cela correspond à une recherche spontanée de stabilité nucléaire. Cette émission de rayonnement accompagne le phénomène de désintégration radioactive, qui transforme le noyau de l'élément "père" (X) en noyau fils (Y). Ainsi, le noyau d'un isotope radioactif va se transformer spontanément en un noyau d'un isotope plus stable du même élément, ou bien encore en un noyau d'un autre isotope plus d'un autre élément chimique ou d’une particule.
La radioactivité naturelle a été découverte par Henri Becquerel (1896) et la radioactivité artificielle par Frédéric Joliot et Irène Joliot-Curie (1934).
3.6.
Les différents rayonnements nucléaires dont les énergies s’échelonnent entre 103 et quelques 106 eV ont différents comportements vis à vis de la matière qu’il rencontre. Ces rayonnements sont assez énergétiques pour ioniser la matière environnante en éjectant des électrons des atomes constituant la matière qui leur est exposée. C'est pourquoi on les considère comme rayonnements ionisants. Ainsi, les radiations nucléaires peuvent endommager les tissus vivants. Cette propriété rend les rayonnements nucléaires (radioactivité) offensifs voire dangereux. Mais cette propriété les conditionne également pour les applications médicales (médecine nucléaire).
Les trois principaux types de radiations nucléaires, α, β et g, pénètrent dans la matière à des degrés différents. Le pouvoir de pénétration, ou mieux le parcours, dépend de la nature du rayonnement dans l’air. En fonction du pouvoir de pénétration, il faut se protéger contre les radiations nucléaires. Pour se protéger contre les effets nocifs des radiations nucléaires, toutes les sources radioactives intenses doivent être entourées par d'épais murs en béton ou un blindage en plomb d'épaisseur différente.

On constate par exemple que les parcours décroissent selon l‘ordre suivant:
Pg >> Pb > Pa
Par voie de conséquence, leur pouvoir ionisant varie en sens opposé: Ia >Ib >>Ig

Le rayonnement α : est le moins pénétrant, car les particules α (ions He2+) sont assez massives et captent des électrons à la surface de la matière pour former des atomes d'hélium. Bien qu'elles ne pénètrent pas très profondément dans un tissu vivant, elles sont très dangereuses, car l'ionisation du tissu humain peut provoquer des troubles graves et des cancers. Ceci est d'autant plus dangereux si les particules α sont inhalées ou ingérées. D'un autre côté, une protection en papier est suffisante pour blinder les rayons α à cause de leur faible pouvoir de pénétration.
Le rayonnement β: est plus pénétrant, car les électrons sont des particules plus petites que les ions d'hélium. Ils peuvent pénétrer jusqu'à environ un centimètre de profondeur dans un tissu vivant avant d'être arrêtés. Ils peuvent être capturés par les bio-molécules qui sont en conséquence ionisées.
Le rayonnement g: est le plus pénétrant de tous les rayonnements nucléaires, car il s'agit de photons de haute énergie, sans charge ni masse qui peuvent traverser la plupart des matériaux. En traversant des tissus vivants comme le corps humain, ils peuvent provoquer des dommages en ionisant des molécules situées sur leur chemin, ce qui peut induire un disfonctionnement de l'ADN et provoquer l'apparition de cancers.
X.7. Activité radioactive
La probabilité de désintégration par unité de temps est donnée par la "constante radioactive" λ et a pour unité l'inverse du temps tel que [λ] =s-1. Cette constante peut être calculée et c’est une caractéristique du noyau radioactif. La constante radioactive varie pour tous les isotopes connus:
![]()
Soit N(0) le stock d'atomes (nombre de noyaux radioactifs présents) d'un isotope radioactif au temps initial, t=0 et soit N(t) , le nombre le plus probable d'atomes radioactifs restant au temps t . N(t) = N(0) ´ e-λt
La "période radioactive" ou de "demi-vie" T½ d'un isotope est le temps moyen qu'il faut attendre pour que 50% du stock de noyaux radioactifs d'un isotope donné soit désintégré:

Nous avons ainsi une relation très important entre la période et la constation radioactive. Les périodes s’expriment dans une unité de temps qui, pour des raisons de commodité peut être évaluée en s, j, années ; elles varient dans des limites énormes selon la nature de l’isotope, c’est ainsi que pour les thoriums, le 218Th a une T/1/2 de 0,18ms, alors que le 232Th (naturel) a une T/1/2 de 1,4*1010 ans (>1017 s), pratiquement 24 ordres de grandeurs

Un radionucléide ayant pénétré dans l'organisme peut soit : se répartir de façon homogène dans tout l'organisme, par exemple : tritium, 24Na, 36Cl; soit se concentrer dans un ou plusieurs organes cibles, par exemple : 131I dans la thyroïde.
Dans le second cas, l'élimination de l'activité incorporée s'effectue par la combinaison de la décroissance radioactive du radionucléide et de l'élimination biologique propre à l'organe cible. En première approximation, on peut considérer que l'élimination biologique obéit à une loi exponentielle de période biologique Tb, qui est le temps nécessaire pour que la moitié de la quantité d'une substance introduite dans un organe en soit éliminée. La demi-vie biologique dépend de l’organe où s’est fixé l’élément considéré. Aux divers organes (os, foie, poumon, etc.) correspondent des constantes de temps biologiques différentes. Ainsi la demi-vie biologique du carbone varie de quelques minutes à 25 jours.
De même, l'élimination biologique d'une contamination interne par un radioélément est caractérisée par une constante d'élimination biologique Tb. Nous avons donc défini une période biologique Tb. C'est le temps au bout duquel la contamination interne se trouve diminuée de moitié c'est-à-dire que la moitié du radioélément se trouve rejetée hors du corps humain.
![]()
En combinant les deux dernières formules, nous pouvons déduire la période effective Te, c'est-à-dire le temps au bout duquel la moitié de la radioactivité du radio-isotope contaminant est éliminée de l'organe critique ou de l'organisme, à la fois par le phénomène de transition spontanée et par les processus physiologiques.



Ce qui signifie que la moitié de l'activité du carbone 14 à l'intérieur du corps disparaît (ou est éliminé) au bout de 12 jours. Malheureusement, ce radio-isotope est renouvelé régulièrement par la chaîne alimentaire.
Loi de la décroissance radioactive
a. Caractère aléatoire d’une désintégration radioactive
- Un noyau instable est susceptible de revenir à l’état stable à tout moment.
- Le phénomène de désintégration est imprévisible. Pour un noyau instable donné, on ne peut prévoir la date de sa désintégration.
- En revanche, on connaît la probabilité de désintégration de ce noyau par unité de temps.
- Le phénomène de désintégration est aléatoire.
- La probabilité qu’a un noyau radioactif de se désintégrer pendant une durée donnée est indépendante de son âge.
- Elle ne dépend que du type de noyaux considéré.
- Un noyau de carbone 14 apparu, il y a mille ans et un autre formé, il y a 5 min ont exactement la même probabilité de se désintégrer dans l’heure qui vient.
- Un noyau ne vieillit pas.
- Ce caractère aléatoire fait que pour un ensemble de noyaux instables identiques, on ne peut prévoir lesquels seront désintégrés à une date donnée, mais on peut prévoir combien de noyaux seront désintégrés.
- On peut prévoir avec précision l’évolution statistique d’un grand nombre de noyaux radioactifs.
- C’est un phénomène sur lequel il est impossible d’agir. Il n’existe aucun facteur permettant de modifier les caractéristiques de la désintégration d’un noyau radioactif.
b. La constante radioactive
Chaque nucléide radioactif est caractérisé par une constante radioactive λ, qui est la probabilité de désintégration d’un noyau par unité de temps, elle s’exprime en s –1. La constante λ ne dépend que du nucléide. Elle est indépendante du temps, des conditions physiques et chimiques. Pendant la durée Δt, la probabilité pour qu’un noyau se désintègre est λ.Δt
c. Loi de la décroissance radioactive
Considérons un échantillon contenant :
- N(t) noyaux radioactifs à la date t.
- A la date t + Δt très proche de t, le nombre de noyaux radioactifs a diminué.
- Pendant l’intervalle de temps Δt très court, on peut considérer que le nombre de noyaux ayant subi une désintégration est : λ.ΔtN.
- La variation ΔN du nombre N de noyaux pendant la durée Δt est donnée par la relation :
- ΔN=-λ.Δt.N soit : ΔN+λ.Δt.N=0 (1)
- Divisons
l’expression (1) par Δt, il vient : ![]()
Lorsque Δt ®
0, l’expression (2) s’écrit : ![]()
La solution de cette équation différentielle du premier ordre donne la loi de décroissance radioactive : N (t) = N 0 e - λ t
Avec :
![]() N 0
représente le nombre de noyaux présent à la date t 0 = 0
N 0
représente le nombre de noyaux présent à la date t 0 = 0
![]() N(t)
représente le nombre de noyaux radioactifs présents à la date t
N(t)
représente le nombre de noyaux radioactifs présents à la date t
![]() λ
est la constante radioactive s –1.
λ
est la constante radioactive s –1.
Énoncé de la Loi de décroissance radioactive :
« Le nombre de noyaux radioactifs N (t) présents à la date t dans un échantillon est donné par la loi de décroissance radioactive ; N (t) = N 0 e - λ t ; N 0 représente le nombre de noyaux radioactifs initialement présents »
d. Demi-vie
Pour un type de noyaux radioactifs, la demi-vie t½ est la durée au bout de laquelle la moitié des noyaux radioactifs initialement présent dans l’échantillon se sont désintégrés
Relation entre t½ et λ :
- Au temps t : N (t) = N 0 e - λ t
- Au temps t + t1/2 : N (t + t 1/2) = N 0 e - λ (t + t ½)
- En conséquence :

Ou ![]()
La demi-vie n’a qu’une valeur statistique. Elle indique qu’un noyau radioactif a une chance sur deux de disparaître au bout d’une demi-vie.
e. courbe de décroissance et constante de temps τ
La constante de temps, notée τ est l’inverse de la constante radioactive. Elle s’exprime en seconde s.
- Expression :
![]()
- On peut obtenir la valeur de la constante de temps τ à partir de la loi de décroissance.

- Si l’on se place au temps t = 0 :

- En conséquence, la tangente à la courbe N (t) = N 0 e - λ t à l’instant initial rencontre l’axe des abscisses à la date τ.
Ou ![]()
L'activité A d'une source radioactive est le nombre de désintégrations par unité de temps.

Remarque: Son unité de mesure est le "Becquerel" est est note [A] = [Bq]. 1 Becquerel correspondant donc à une désintégration par seconde. L'ancienne unité de mesure de la radioactivité était le "Curie" [Ci].
![]()
la désintégration par seconde (dps): 1 Bq = 1 dps , la désintégration par minute (dpm) : 1 dps correspond à 60 dpm . Le nombre de coups par minute (cpm) : mesure de la radioactivité effectuée par un compteur de radioactivité.


Example 1: Le cycle naturel de formation du carbone 14 fait qu'en moyenne le carbone présent dans la matière végétale vivante a une activité de 15,3 désintégrations de 14C par minute et par gramme de carbone. L'étude des célèbres peintures des grottes de Lascaux (France) montre que le charbon de bois qui y fut utilisé manifeste 2,22 désintégrations par minute et par gramme de carbone. En déduire l'âge de ces fresques et évaluer l'incertitude de cette détermination.
SOLUTION

Example 2:Les batteries d'un "pacemaker" contiennent du plutonium-238 dont le temps de demi-vie est de 27,1 années. Si l'échantillon initial contient 2,57 109 atomes de Pu, combien de temps s'écoulera avant que le nombre d'atomes soit réduit à 5,02 108 atomes ? Réponse : La variation de l'activité radionucléaire est donnée par la relation
N = No e -lt
ou encore en remplaçant la constante radioactive l par la période T de l'élément
N = No e - 0,693 t / T puisque l = 0,693 / T

Example 3 : Le sodium-24 est utilisé en médecine pour contrôler la circulation sanguine. Sa demi-vie est de 15 heures. Quel est le pourcentage de cet isotope encore présent dans un échantillion après 60 h. Réponse : 6,25 %
Example 4 : Le polonium-212 subit une désintégration avec émission d’une particule alpha. Quel est l’élément produit. Réponse : plomb-208
Example 5 : Un objet contient 12,5% de l’abondance actuelle en carbone-14. Quel est l’âge de cet objet. Réponse : 17200 ans.
Chapitre 4ème : Nomenclature
4.1. Eléments
Les noms des éléments doivent être connus. Ils sont symbolisés dans une équation chimique par leur symbole. Ce dernier est composé d’une ou deux lettres, généralement les premières lettres du nom, la première lettre étant majuscule et la deuxième minuscule. Exemple C pour carbone, Fr pour francium, S pour soufre. Il se peut que le symbole de l’élément corresponde à son nom dans une langue autre que le français. Exemple Na pour sodium (natrium en latin) et W pour tungstène (wolfram en allemand).
4.2. Composés binaires de type I (ioniques)
Les composés binaires ioniques contiennent un ion positif (cation), toujours écrit le premier dans la formule chimique, et un ion négatif (anion).
Pour nommer ces composés, les règles suivantes s’appliquent :
1. l’anion est toujours nommé en premier et le cation en second
2. un cation monoatomique (d’un seul atome) a le même nom que son élément. Par exemple,
Na+ est appelé sodium dans les composés contenant cet ion. Le calcium est Ca2+. Lorsqu’un element metallique peut donner lieu à deux cations differents, la charge sur l’ion métallique doit être spécifiée en chiffres romains. Ces derniers indiquent le nombre d’oxydation après le nom du metal. Cu +1 : le cation cuivre (I), Cu + 2 : le cation cuivre (II).
Les cations monoatomiques non-metalliques sont nommés à partir des noms des éléments, en remplacant la dernière (ou les deux dernières) syllabe(s) par le suffixe onium.
Cl + chloronium , Br+ bromonium et NH4+ ammonium.
3. un anion monoatomique est nommé en prenant la première partie du nom de l’élément et en rajoutant le suffixe -ure (sauf si c’est de l’oxygène, auquel cas le suffixe est -ide). Donc l’ion Cl- est appelé chlorure. Le NaCl est donc le chlorure de sodium, BaS : sulfure de baryum, Mg3N2: nitrure de magnesium, ZnS : sulfure de zinc, LiH: hydrure de lithium, Ag3P: phosphure d’argent, CaO : oxyde de calcium, CrO3 : oxyde de chrome (VI), Cr2O3 : oxyde de chrome (III), CrO : oxyde de chrome (II), Na2O : oxyde de sodium, Na2O2 : peroxyde de sodium. Inversément le bromure de césium a pour formule chimique : CsBr
4.3. Composés binaires ioniques de type II
Dans les composés binaires de type I, le métal impliqué ne forme qu’un type de cation. Le sodium ne forme que du Na+, le calcium du Ca2+, et ainsi de suite. Cependant, il y a beaucoup de métaux qui peuvent former plus d’un type d’ion positif et peuvent donc former plus d’un type de composé ionique avec un anion donné. Par exemple, le composé FeCl2 contient des ions Fe2+, et le composé FeCl3 contient des ions Fe3+. Dans un cas comme cela, la charge sur l’ion métallique doit être spécifiée. Les noms systématiques pour ces deux composés du fer sont chlorure de fer (II) et chlorure de fer (III), respectivement, où les chiffres romains indiquent la charge du cation.
Un autre système de nomenclature pour ces composés existe dans la littérature plus ancienne. Pour les métaux qui ne peuvent donner que deux ions, l’ion avec la charge la plus élevée a un nom suivi du suffixe -ique, alors que celui avec la charge la plus petite a un nom avec le suffixe -eux. Dans ce système, Fe3+ est le cation ferrique, Fe2+ est le cation ferreux. Les noms de FeCl3 et de FeCl2 sont donc respectivement chlorure ferrique et chlorure ferreux.
L’utilisation de chiffres romains est de mise seulement quand plus d’un composé ionique peut être formé a partir d’une paire donnée d’éléments. Cela se produit le plus souvent pour les composés contenant des métaux de transition, qui forment souvent plus d’un cation. Les éléments qui ne forment qu’un cation ne doivent pas être identifiés par un chiffre romain. Les éléments des groupes IA et IIA, ainsi que l’aluminium, ne nécessitent pas de chiffres romains.
4.4. Composés ioniques avec ions polyatomiques
Nous n’avons pas encore considéré les composés ioniques qui contiennent des ions polyatomiques. Par exemple, le composé nitrate d’ammonium, NH4NO3, contient les ions polyatomiques NH4+ et NO3 -. Les ions polyatomiques ont des noms spéciaux qui doivent être mémorisés pour pouvoir nommer les composés qui les contiennent. Les ions polyatomiques les plus importants sont repris dans le tableau suivant :

Nous remarquons par ailleurs que plusieurs anions contiennent un nombre différent d’atomes d’oxygène (oxanions). Quand il y a deux possibilités, l’anion avec le plus grand nombre d’oxygènes reçoit le suffixe -ate, celui avec le plus petit nombre, le suffixe -ite. Quand plus de deux oxanions existent pour une série, on rajoute les préfixes hypo (pour le plus petit nombre d’oxygène) et per- (pour le plus grand nombre d’oxygènes).
4.5. Composés binaires de type III : covalents - contenant deux non métaux
Les composés binaires covalents sont formés par deux non métaux. Bien qu’ils ne contiennent pas d’ions, leur nomenclature ressemble à celle des composés ioniques :
1. le second élément de la formule est nommé en premier, avec le même suffixe que pour les anions càd en rajoutant le suffixe -ure au nom de l’anion (sauf si c’est de l’oxygène, auquel cas le suffixe est -ide)
2. le premier élément est nommé en second
3. des préfixes (mono, di, tri, tetra…) sont rajoutés au premier nom pour indiquer le rapport stoechiométrique entre les éléments si nécessaire.
Exemple : CO2 est le dioxyde de carbone, CO est le monoxyde de carbone, Na2S : sulfure de disodium ou sulfure de sodium, SF6 : hexafluorure de soufre, MnO2 : dioxyde de manganese, ICl3 : trichlorure d’iode, NO2 : dioxyde d’azote, ClO3 : trioxyde de chlore.
Certains composés sont toujours appelés par leurs noms usuels et non leur nom systématique. L’eau et l’ammoniac sont les meilleurs exemples. Les noms systématiques de H2O et NH3 ne sont jamais utilisés.Il en est de meme H2O2 : eau oxygenee, H2N-NH2 : hydrazine, CH4 : methane, SiH4 : silane, SbH3 : stibine….
4.6. Acides
Lorsqu’elles sont dissoutes dans l’eau, certaines molécules produisent une solution contenant des ions H+ libres (protons). Ces substances, les acides, possèdent leur propre nomenclature. Les règles dépendent si l’anion contient ou non de l’oxygène. Si l’anion ne contient pas d’oxygène, l’acide est appelé « acide Xxhydrique » ou Xx est la racine du nom de l’anion. HCl est donc l’acide chlorhydrique, H2S est donc l’acide sulfuhydrique, H2Se est donc l’acide sélénhydrique
Quand l’anion contient de l’oxygène, le nom de l’acide est formé par la racine du nom de l’ion suivie du suffixe -ique ou -eux. Si le nom de l’anion correspondant se termine par ate, alors le suffixe sera -(r) ique, si l’anion se termine par -ite, alors le suffixe sera -eux.
H2SO4 est donc l’acide sulfurique, H2SO3 : acide sulfureux, H2S2O8 : acide persulfate, HNO3 : acide nitrique et HNO2 : acide nitreux, H2CO3: acide carbonique et H2SiO3: acide silicique.
S’ils existent plus de deux oxacides avec le meme atome central, on fait intervenir les prefixes hypo- et per- . HClO : acide hypochloreux, HClO2 : acide chloreux, HClO3 : acide chlorique, HClO4 : acide perchlorique. HIO : acide hypoiodeux, HIO2 : acide iodeux, HIO3 : acide iodique, HIO4 : acide periodique. H3PO4: acide phosphorique, H3PO3 : acide phosphoreux, H3PO2 : acide hypophosphoreux. H2MnO3: acide manganeux, H2MnO4: acide manganique et HMnO4: acide permanganique.
Remarques:
Le préfixe thio indique qu’un atome de soufre remplace totalement ou partiellement un atome d’oxygène dans une molécule. Example : H2S2O3 : acide thiosulfurique, H2CS3 : acide trithiocarbonique.
Les acides thioneux et thioniques se caracterisent par la présence de liaisons S-S. Example : H2S2O2 : acide dithioneux, H2S2O6 : acide dithionique, H2S4O6 : acide tétrathionique.
4.7. Bases
Les bases sont des composes qui se dissocient en solution aqueuse en ion metallique et en ion hydroxyde. NaOH : hydroxyde de sodium, Ca (OH)2 : hydroxyde de calcium, Al(OH)3 : hydroxyde d’aluminium, Fe(OH)2 : hydroxyde de fer (II) ou hydroxyde ferreux , Fe(OH)3 :hydroxyde de fer (III) ou hydroxyde ferrique, Mn(OH)2 : hydroxyde de manganèse.
4.8. Sels
Les sels sont des composés obtenus en remplacant les hydrogènes d’un acide par des atomes métalliques. On parle des sels neutres lorsque tous les hydrogènes acides ont été substitués, par example Na2SO4 : sulfate de sodium, Na3PO4 : phosphate de sodium, KClO : hypochlorite de potassium, KClO4 : perchlorate de potassium, K2WO4 : tungstate de potassium, Na2CrO4 : chromate de sodium, KMnO4 : permanganate de potassium.
Il existe aussi les sels hydratés càd contenant de l’eau dans leurs formules et on les nomme de la manière suivante : CuSO4.5H2O : sulfate de cuivre (II) pentahydraté, Na2SO4.10H2O : sulfate de sodium décahydraté, MgSO4.7H2O : sulfate de magnesium heptahydraté, CaSO4.2H2O : sulfate de calcium dihydraté et K Al (SO4)2.12H2O : sulfate de potassium et d’aluminium dodecahydraté.
Lorsque le remplacement des atomes d’hydrogènes par les atomes métalliques est incomplet, on obtient un sel acide (hydrogenosel) par exemple NaHSO4:bisulfate de sodium ou hydrogenosulfate de sodium, MgHCO3 : bicarbonate de magnesium, NaHCO3 : bicarbonate de sodium, KHS : bisulfure de potassium ou hydrogeno sulfure de sodium, NaH2PO4 : dihydrogenophosphate de sodium.
4.9. Des noms aux formules
Le passage des noms aux formules est tout aussi important, et il faut tenir compte des règles énoncées ci-dessus pour retrouver les formules correspondantes. La formule empirique d'un composé est son écriture dans le language chimique. C'est la formule qui interviendra dans les équations chimiques. Etablissons la formule de l'hydroxyde de calcium : Hydroxyde signifie que le groupement OH- est présent. Calcium signifie que nous aurons l'ion Ca2+ (celui ci n'a qu'un seul état d'oxydation car rien n'est précisé dans la formule). La molécule doit être neutre, donc nous devons avoir deux anions OH- par ion Ca2+. La formule est donc Ca (OH)2.
Chapitre 5ème : Réaction Chimique : Stoechiométrie
Parmi les quatre conditions fondamentales de la vie (alimentation, isolation et échange avec le monde extérieur, contrôle des températures internes et reproduction), les trois dernières sont entièrement subordonnées aux transformations de la matière vivante par des réactions chimiques ou transformations qui permettent de produire des éléments de construction (os, muscles, tissus, membres, etc. …) et l'énergie nécessaire à leur mise en place et à leur fonctionnement. La réaction chimique est indispensable pour comprendre les phénomènes vitaux.
Une réaction chimique correspond à la transformation de réactifs en produits. L'équation chimique établit les rapports qualitatifs et quantitatifs associés à la transformation considérée:
![]()
La flèche indique le sens du déroulement de la réaction. Une double flèche indique que la réaction est terminée et a atteint l'équilibre chimique.
L'équation chimique d'une réaction correspond à une transformation microscopique impliquant des atomes et des molécules.
Exemples:
![]()
Les nombres qui multiplient les formules chimiques entières dans l’équation sont appelés
coefficients stoechiométriques. Ceux-ci indiquent les nombres relatifs de moles de réactifs et des produits qui participent à la réaction.
L’équation peut également donner l’état physique des réactifs et des produits, en utilisant les abbréviations (s) pour solide, (l) pour liquide, (g) pour gaz et (aq) pour solution aqueuse (aqueux signifie que le composé est dissout dans une solution aqueuse):
![]()
5.1.
L’équilibrage de l’équation chimique (l’ajustement des coefficients stoechiométriques) à partir de l’équation qualitative doit prendre en compte:
a) la conservation du nombre de chacun des atomes impliqués (en l’absence de réaction nucléaire, les atomes ne sont ni créés ni détruits pendant la réaction)
b) la conservation de la charge électrique totale et donc du nombre d’électrons.
Il existe cependant plusieurs méthodes rigoureuses dont celle décrite ci-dessous qui est illustrée pour la réaction de combustion du propane C3H8.
1/ Formuler l'équation non-équilibrée ou équation squelette qui résume l'information qualitative de la réaction:
![]()
2/ Introduire les coefficients stoechiométriques sous forme d'inconnues a, b, c, d:
![]()
3/ Equilibrer l'équation revient à déterminer a, b, c et d en effectuant le bilan du nombre d'éléments présents dans chaque membre de l'équation:
C : 3a = c, H : 8a = 2d, O : 2b = 2c + d
4/ On obtient 3 équations et 4 inconnues. Il suffit de considérer un coefficient égal à 1 pour l'élément le moins fréquent, soit dans l'exemple décrit le carbone ou l'hydrogène; on choisira le carbone, donc a = 1. Il s'en suit que c = 3, d = 4, b = 5.
5/ L'équation est maintenant équilibrée:
![]()
Dans le cas de réactions d’oxydo-réduction (échange d’électrons) un bilan des degrés d’oxydation des éléments est à prendre en compte pour satisfaire la conservation de la charge. Ce concept sera introduit plus loin.
Exemple : Equilibrer l’équation chimique suivante:
Al 3+ + NO3– + H2O + OH– [Al(OH)4] – + NH3
Réponse:
On pose l’équation avec des coefficients alphanumériques

5.2.
5.2.1. Calculs stoechiométriques : quantité de réactifs et produits
Les coefficients stoechiométriques représentent un nombre de molécules, et non des masses. Cependant, dans un laboratoire ou une usine, les quantités de substances demandées ne sont jamais exprimées pratiquement en molécules, elles le sont en masse, volume,... Nous voyons ici comment utiliser une équation chimique pour déterminer les masses des réactifs et produits.
Prenons l'exemple de la réaction du propane avec de l'oxygène qui produit du dioxyde de carbone et de l'eau. Quelle masse d'oxygène va réagir avec 98.1 g de propane.
La première chose à faire est d'écrire l'équation chimique balancée correspondante :
C3H8 + 5O2 3CO2 + 4H2O
Cette équation signifie que 1 mole de propane réagit avec 5 moles d'oxygène pour former 3 moles de dioxyde de carbone et 4 moles d'eau. Ces proportions sont toujours respectées dans la réaction chimique. Combien de moles de propane représentent 98.1 g ?
1 mole de propane pèse 3x12.01 g C + 8x1.008 g H = 44.1 g
Donc 98.1 g représente 98.1/44.1 = 2.18 mole
Maintenant nous revenons au fait que 1 mole de propane doit réagir avec 5 mole d'oxygène, donc 2.18 mole de propane doivent réagir avec 5x2.18 = 10.9 mole d'oxygène (le rapport de 5 doit être respecté. Finalement nous savons qu'une mole d'oxygène (O2) pèse 32.0 g, donc 10.9 mole pèsent 32.0 x 10.9 = 349 g d'O2. Nous pouvons étendre la question à : quelle masse de dioxyde de carbone sera formée par la combustion de ces 98.1 g de propane ? Nous savons (voir équation) que 1 mole de propane donnera, après réaction, 3 moles de CO2, donc, 2.18 mole donneront 3x2.18=6.54 mole de CO2. Comme la masse molaire du CO2 est de 44.0 g, nous formerons 6.54 x 44.0 g = 288 g de CO2.
5.2.2. Etapes pour calculer les masses de réactifs et produits dans une réaction chimique :
0. écrire l'équation chimique correspondant à la réaction décrite
1. balancer l'équation de la réaction chimique
2. convertir les masses de réactifs ou produits en mole de substance
3. utiliser l'équation balancée pour employer le bon rapport de moles
4. utiliser le rapport de moles pour calculer le nombre de moles du réactif ou produit désiré
5. convertir les moles en grammes si demandé dans le problème.
5.2.3.Exercices
1. l'hydroxyde de lithium solide est utilisé dans la navette spatiale pour retirer le dioxyde de carbone de l'environnement et le transformer en carbonate de lithium solide et eau liquide. Quelle masse de dioxyde de carbone peut être absorbée par 1.00 kg d'hydroxyde de lithium.
2. Le bicarbonate de sodium (NaHCO3) est utilisé comme antiacide. Il neutralise l'excès d'acide chlorhydrique produit dans l'estomac. Les produits de la réaction sont du chlorure de sodium, de l'eau et du dioxyde de carbone. D'autre part, le lait de magnésie (qui est une solution aqueuse d'hydroxyde de magnésium) est aussi utilisé comme antiacide. Sa réaction avec l'acide chlorhydrique forme de l'eau et du chlorure de magnésium (MgCl2). Quel est l'antiacide le plus efficace par gramme ?
5.2.4. Calculs impliquant un réactif limitant
Quand des réactifs sont mélangés pour effectuer une réaction, ils le sont souvent dans des quantités stoechiométriques, c'est à dire, dans les quantités correctes telles que définies au point précédent, qui font que tous les réactifs sont consommés entièrement en même temps. Néanmoins, dans beaucoup d'autre cas, les quantités de réactifs présents ne respectent pas les proportions stoechiométriques. Un des réactifs va, par sa quantité, limiter la réaction. Le réactif limitant d'une réaction est l'espèce pour laquelle la quantité disponible est inférieure à celle exigée par la stoechiométrie de la réaction.
Prenons l'exemple suivant :
L'azote gazeux peut être préparé en passant de l'ammoniac gazeux sur de l'oxyde de cuivre (II) à haute température. Les autres produits de réaction sont du cuivre métallique et de la vapeur d'eau. Si un échantillon contient 18.1 g d'ammoniac réagit avec 90.4 g de CuO, combien de grammes d'azote peut on former, et quel est le réactif limitant ?
L'équation chimique correspondante est :
2NH3 + 3CuO N2 + 3Cu +3H2O
Le nombre de mole d'ammoniac (masse molaire 17.03 g/mol) est de 18.1 g/17.03g/mol =1.06 mol. Le nombre de mole de CuO est de 90.4/79.55=1.14 mol Pour déterminer le réactif limitant, nous utilisons le rapport de moles entre CuO et NH3 dans l'équation :
1.06 mol ammoniac x 3 mol CuO/2mol NH3= 1.59 mol CuO. Donc il faut 1.59 mol CuO pour réagir avec 1.06 ammoniac. Comme nous n'avons que 1.14 mol à notre disposition, CuO est le réactif limitant. La quantité d'azote formé dépend donc de la quantité de CuO présent. Comme dans l'équation nous avons 1 mole d'azote formée pour 3 moles d'oxyde de cuivre, et que nous avons en réalité 1.14 mole CuO, on formera donc : 1.14 x 1/3 = 0.380 mole d'azote gazeux. En utilisant la masse molaire de l'azote (28.0 g/mol), nous trouvons la masse d'azote formée : 0.380 x 28.0 = 10.6 g d'azote.
La connaissance d'une réaction chimique équilibrée permet de faire des prédictions quant aux quantités de produits formés à partir d'une masse de réactifs à disposition et, inversement, de déterminer une masse de réactifs mis en jeu, connaissant la quantité de produits formés.
Le réactif limitant d'une réaction est l'espèce pour laquelle la quantité disponible est inférieure à celle exigée par la stoechiométrie de la réaction.
Equation équilibrée de la formation de l'acétylène à partir du carbure de calcium:
![]()
Expérience : Lorsque l'on verse 100 g d'eau sur 100 g de carbure de calcium, il se forme de l'hydroxyde de calcium et de l'acétylène. Pour identifier le réactif limitant, on procède selon cette méthode (voir cours théorique)
Une équation chimique ne correspond qu'à un bilan de masse et ne décrit pas forcément le mécanisme de cette réaction. La grande majorité des réactions chimiques ont lieu en une série d'étapes, chacune correspondant à une réaction élémentaire:

Un produit de la première étape, X, qui est éliminé lors de la deuxième étape, est appelé intermédiaire réactionnel car il n'apparaît pas dans la stoechiométrie de cette réaction (il peut, dans certains cas, être isolé ou observé pendant la réaction).
Le processus détaillé par lequel une réaction complexe a lieu est décrit par le mécanisme de la réaction. En biochimie, les réactions compliquées peuvent comprendre des dizaines d'étapes successives.
5.2.5. Classification des réactions chimiques
Il existe des milliards de réactions possibles, et le chimiste les groupe en différentes catégories. les réactions sont divisées en les groupes suivants :
- Réactions de précipitation
- Réactions acide base
- Réactions d'oxydoréduction
Quasiment toutes les réactions peuvent être classées dans une de ces trois catégories. Nous reviendrons plus loin sur ces différents types de réaction. Nous allons ici décrire seulement la stoechiométrie de ces réactions, ainsi que leurs particularités, afin de permettre la résolution des différents problèmes.
a) Réactions de précipitation
Quand un produit insoluble est produit par la réaction de deux solutions d’électrolytes, on parle de précipitation.
Exemples:
BaCl2 (aq) + K2SO4 → BaSO4 (s) + 2 KCl (aq)
Cd(NO3)2 (aq) + Na2S (aq) → CdS (s) + 2 NaNO3 (aq)
Dans le cas d’électrolytes forts, l’équation peut être écrite sous la forme ionique complète:
Cd2+ + 2NO3- + 2Na+ + S2- → CdS + 2Na+ + 2NO3-
Ici les ions Na+ et NO3– apparaissent de part et d’autre de l’équation. Ce sont des ions spectateurs. L’équation peut alors être simplifiée sous une forme ionique réduite:
Cd2+ + S2- →CdS↓
b) Réactions acide-base
La réaction entre un acide et une base est appelée réaction de neutralisation. Le composé ionique produit par la réaction est appelé un sel :
Acide + base → sel + eau
HCl (aq) + NaOH (aq) → NaCl (aq) + H2O (l)
H2SO4 (aq) + Mg(OH)2 → MgSO4 (aq) + 2 H2O (l)
Chaque ion hydroxyde réagit avec un ion hydronium. L’équation ionique réduite de la dernière réaction est: 2 H3O+ (aq) + 2 OH– → 4 H2O (l),
ou encore H3O+ (aq) + OH– → 2 H2O (l)
Elle est la même pour toute réaction de neutralisation d’un acide par une base formant un sel soluble. Dans certains cas, la réaction de neutralisation aboutit à la formation d’un gaz:
CaCO3 (aq) + 2 HCl → CaCl2 + H2CO3 (aq)
H2CO3 (aq) → H2O (l) + CO2 (g)
c) Réactions d’oxydo-reduction
L’oxydation est une transformation chimique dans laquelle des électrons sont perdus par un atome, une molécule ou un ion. La réduction est une transformation chimique dans laquelle des électrons sont gagnés par un atome, une molécule ou un ion.
Exemples:
Fe → Fe2+ + 2 e– les atomes de Fe sont oxydés en ions ferreux Fe2+
Cl2 + 2 e– → 2 Cl– le chlore moléculaire est réduit en chlorures
Comme le nombre total d’électrons doit être conservé, l’oxydation et la réduction ont toujours lieu simultanément. On parle de réaction d’oxydo-réduction (ou de réaction rédox): le nombre total d’électrons perdus par oxydation doit être égal au nombre d’électrons gagnés par réduction.
Nombre d’oxydation (NO)
Définitions
Le degré d’oxydation ou nombre d’oxydation d’un atome dans une combinaison chimique est une charge arbitraire assignée à l’atome à partir de certaines règles. L’état ou le degré d’oxydation d’un élément est caractérisé par un nombre d’oxydation (NO). C’est un nombre entier positif, négatif ou nul. Ce nombre indique l’importance de la perte ou du gain d’électrons de l’élément, par rapport à l’atome neutre.
1) Dans les composés ioniques binaires, le degré d’oxydation est égal à la charge par atome.
2) Dans les composés covalents, les électrons participant à la liaison ne sont pas complètement transférés d’un atome à l’autre mais se partagent entre les atomes liés. Toutefois, par convention, on attribue chaque électron de la liaison à un atome particulier:
– Si les atomes sont identiques, on attribue la moitié des électrons de la liaison à chacun des atomes.
– Si les atomes sont différents, tous les électrons de la liaison sont attribués arbitrairement à celui des atomes qui possède la plus grande électronégativité. L’électronégativité est définie comme étant la propriété d’un atome à attirer à lui les électrons d’une liaison. Plus la différence d’électronégativité sera grande entre deux atomes liés et plus la liaison sera polarisée et aura un caractère ionique. Les éléments les plus électronégatifs dans l’ordre d’électronégativité décroissante sont F > O > N > Cl. Le carbone C est plus électronégatif que H. Les métaux sont moins électronégatifs que les non-métaux.
.Détermination du nombre d’oxydation
Règles permettant de déterminer le NO d’un élément
i) On attribue aux atomes à l'état élémentaire (élément non combiné à un autre) le degré d'oxydation zéro : NO = 0 (exemple: Na ; Al : NO = 0). Il en est de même pour les combinaisons neutres formées d’un seul élément : NO = 0 (exemple: Br2 ; O2 : NO = 0)
ii) Le degré d'oxydation des métaux alcalins (Li, Na, K, Rb et Cs) inclus dans des composés est toujours égal à +1. Le degré d’oxydation des métaux alcalino-terreux (Be, Mg, Ca, Sr, Ba et Ra) ainsi que de Zn et Cd, inclus dans des composés est égal à +2.
iii) Ions monoatomiques : le NO = charge portée par l’ion (exemple: S2- (-II) ; Sn4+ (+IV))
iv) La somme des degrés d'oxydation de chacun des atomes présents dans une molécule est égale à zéro. S'il s'agit d'un ion, la somme des degrés d'oxydation est égale à la charge du ion (exemple: H2O : 2 NO (H) + NO (O) = 0 et MnO4- : NO (Mn) + 4 NO (O) = -1).
v) Le fluor (élément le plus électronégatif) inclus dans un composé a le degré d’oxydation de -1 (sauf dans F2 : NO = 0),
vi) L'hydrogène présent dans un composé a le degré d’oxydation +1 (sauf s'il s'agit d'un hydrure où il est à -1 par exemple dans NaH ou CaH2). L'oxygène (élément le plus électronégatif après le fluor) a le degré d'oxydation -2 sauf s'il s'agit de peroxydes (–O-O–), où il vaut -1(-1/2 dans les superoxydes, comme par ex. dans KO2) ou dans les composés fluorés où il peut être positif.
Exemple : F2O F (- I)
Soit x le nombre d’oxydation de l’oxygène dans F2O : 2 (-I) + x = 0, x = +II dans F2O
H2O2 : 2 (+I) + 2x = 0 x = -I
vii) Le NO de H = +I sauf dans H2 : NO = 0 et dans les hydrures NaH : NO(H) = -I
viii) Dans les molécules complexes (en particulier les molécules organiques) un même élément peut être présent dans différents états d’oxydation. Le nombre d’oxydation est alors le nombre d’électrons virtuellement "donnés" ou "captés" par un atome lié en comparaison avec son état fondamental. Ce nombre est compté négativement lorsque l’atome considéré est plus électronégatif que l’atome auquel il est lié, positivement dans le cas contraire et nul si les deux atomes sont identiques.


L’augmentation du degré d’oxydation représente une oxydation. La diminution du nombre d’oxydation représente une réduction.
Exemples
Fe + Cl2 → Fe2+ + 2 Cl–
MnO2 + 4 HCl → MnCl2 + Cl2 + 2 H2O
Le degré d’oxydation du Fe passe de 0 à +2 (on note que Fe0 a été oxydé en FeII). Celui du Cl passe de 0 à –1 (Cl0 a été réduit en Cl-I ). Le degré d’oxydation du manganèse dans
MnO2 est +4. Il est réduit à +2 dans Mn2+ (MnIV réduit en MnII). Celui de l’oxygène dans MnO2 et H2O reste inchangé à –2.
Exercices
1 a) Déterminer le degré d’oxydation de l’azote dans chacune des substances suivantes : N2O ; NO ; HNO3 ; NH3 .
b) Donner la formule de Lewis des molécules de NO et de NH3 et déterminer leur géométrie.
c) Indiquer la polarité des liaisons N–O et N–H à l’aide d’une flèche.
Réponse :
a) Comme :
– La somme des degrés d’oxydation des atomes du composé est zéro.
– Le degré d’oxydation d’hydrogène est +1 en combinaison avec les non-métaux.
– Pour l’oxygène le degré d’oxydation est -2 dans la plupart des composés.
Donc le degré d’oxydation de l’azote dans leurs composés suivants sont : N2O ( +1); NO (+2) ; HNO3 (+5); NH3(–3)
b) La configuration électronique à l’état fondamental des éléments :
N : 1s2 2s2 2p3 : la couche de valence
O : 1s2 2s2 2p4 : la couche de valence
H : 1s1
Les formules de Lewis des molécules formées :

c) La dissymétrie dans la répartition électronique entre les 2 atomes crée un dipôle électrique d’autant plus marqué que la différence d’électronégativité est grande. Les électronégativités des éléments : EN ; O : 3,44 ; N : 3,04 ; H : 2,20 ainsi Δ EN = 0,3 (NO) et Δ EN = 0,84 (NH3)

Chapitre 6ème : Les Equilibres Chimiques
6.1. Introduction
Une réaction chimique ne se traduit pas toujours par la disparition complète du réactif minoritaire ; de nombreuses réactions sont partielles et aboutissent à un équilibre entre les réactifs de départ et les produits de la réaction.
Dans une réaction de combustion, comme celle du propane avec l'oxygène, la réaction s'arrête lorsque l'un des deux réactifs est totalement épuisé ; ce type de réaction est qualifié de réaction totale, complète ou irréversible. Une réaction chimique ne se traduit pas toujours par la disparition complète du réactif minoritaire ; de nombreuses réactions sont partielles et aboutissent à un équilibre entre les réactifs de départ et les produits de la réaction ; ce type de réaction est qualifié de réaction réversible, incomplète ou inversible1.
Guldberg et Waage (1865), en s'inspirant de propositions de Berthelot, ont montré empiriquement qu'il existait une relation entre les concentrations des espèces présentes à l'équilibre en solution. La constante d’équilibre, relative aux concentrations, a été appelée constante de Guldberg et Waage ou constante de la loi d'action de masse.
Le principe de Le Chatelier établi empiriquement en 1884 indique également l'évolution d'un équilibre lors d'une modification des conditions de la réaction. Ce principe dit « de modération » stipule qu'un équilibre s'oppose aux changements extérieurs qui tentent de le modifier. Il existe cependant des cas dans lesquels ce principe n'est pas vérifié.
Le développement ultérieur de la thermodynamique par Willard Gibbs, Théophile De Donder et Gilbert Lewis, et l'application de la fonction enthalpie libre aux réactions chimiques effectuées à température et pression constantes, ont permis de démontrer les relations formulées empiriquement par Guldberg et Waage, et Le Chatelier.
Pour cela, il est nécessaire de définir précisément des grandeurs de réaction indispensables à la compréhension des phénomènes : avancement de réaction , enthalpie libre de réaction , enthalpie libre standard de réaction , quotient de réaction et constante d'équilibre . Ces outils permettent de prévoir le sens d'une réaction, le positionnement de l'équilibre et la composition du système.
6.2 Notion de Concentration
La concentration d'un soluté dans une solution décrit la quantité du dit soluté qui est dissoute dans une quantité déterminée de solvant, voire de solution.
D’où une solution est une phase homogène qui est constitué d’un solvant et d’une solution.
En chimie analytique, on utilisera souvent les solutions aqueuses, celle ayant comme solvant l’eau distillée.
Les expressions de la concentration
1. Le titre d’une solution (T)
C’est la masse en gramme du soluté dans 1ml de solution ;
![]()
2. Le pourcentage (%)
Le pourcentage en masse d’une solution c’est la masse du soluté contenue dans 100 g de solution ;
![]()
Tandis qu’un autre pourcentage en volume d’une solution est le volume du soluté contenu dans 100 ml de solution.
![]()
3. La molarité (M)
La manière la plus couramment employée pour exprimer la concentration d'un soluté est de parler de sa molarité, qui est représentée par le symbole M. La molarité est définie comme étant le nombre de moles d'un soluté qu'il y a par litre de solution :
Molarité=![]()
D’où ![]()
Où M représente la molarité de la solution, n est le nombre de moles du soluté dissous dans la solution et V est le volume de la solution exprimé en litres.
Quelque formule usuelle :
![]()
Avec : m : la masse de la substance dans l’échantillon
Mm : la masse molaire
V : le volume
4. La molalité (M’)
C’est le nombre de mole de soluté contenu dans 1000g de solvant (eau)
![]()
Elle s’exprime en g/1000g
5. La normalité
C’est le nombre d’équivalent gramme contenu dans une litre de solution.
![]()

Balance"massique".
111 g de chlorure de calcium se répartissent en 71 g de chlorures et 40 g de
calcium : il n'y a pas équilibre des quantités massiques des anions et des
cations.
Par exemple nous avons vu qu'une solution de 20 mg.l-1 de calcium est une solution dont la concentration molaire est : [Ca2+] = 0,5 mmol.L-1. Le nombre de moles de charges électriques élémentaires est : 2x0,5 = 1mmol.L-1. On peut alors écrire que la concentration en calcium est 1 mmol.L-1 de charges électriques élémentaires.
Si l'on définit une unité appelée équivalent telle que : l'équivalent est égal à 1 mole de charges électriques élémentaires, l'écriture peut être simplifiée en écrivant :
[Ca2+] = 10-3 eq.L-1.
On définira le milli-équivalent comme étant égal à 1 mmol de charges électriques élémentaires.
La
masse nécessaire pour porter 1 équivalent est fonction de l'espèce ionique.
Cette masse s'exprime par la relation :![]() avec
: Mm en g.mol-1
avec
: Mm en g.mol-1
et x en eq.mol-1 et est appelée équivalent-gramme.
x : valence de l'ion (unité :
nombre de moles de charges électriques élémentaires portées par une mole d'ion
: soit avec la notation proposée eq.mol-1 ),
Mm : masse molaire de l'ion.
Exemple
: un équivalent-gramme d'ion sulfate vaut : ![]() =48 grammes par équivalent.
=48 grammes par équivalent.
Pour passer des concentrations massiques aux concentrations exprimées en quantité de charges électriques (eq.L-1) il faut donc réaliser deux opérations :
1. Déterminer l'équivalent-gramme
de chaque espèce.
2. Diviser la concentration massique par l'équivalent-gramme.
Ex : Equivalent-gramme du calcium : 20 mg.meq-1 ; Concentration massique : [Ca2+] =100 mg.L-1 alors : [Ca2+] = 100/20 = 5 meq.L-1.
Remarque : une solution normale contiendra un équivalent de soluté par litre de solution. Une solution décinormale contiendra un dixième d'équivalent par litre, ...
Exemples :
Solution normale d'acide chlorhydrique : 36,5 g.L-1 d'HCl (masse molaire : 36,5
g.mol-1, valence : 1).
Solution normale d'acide sulfurique : 49 g.L-1 (masse molaire : 98 g.mol-1,
valence : 2).
Formules usuelles :
![]()
Avec : m : masse de la substance
néq : le nombré d’équivalent gramme
Méq : la masse équivalente
x : le nombre d’ion échangé
Mm : la masse molaire
La détermination de x dépend d’un équilibre chimique à un autre : d’où
- Pour les acides x est le nombre d’ion H+, participant à la réaction ou présent dans la molécule d’acide :
Ex : ![]()
Cela dépend aussi de la force de dissociation de l’acide.
- Pour les bases x est le nombre d’ion OH-, participant à la réaction ou présent dans la molécule de la base
![]()
![]()
- Pour les sels x est le nombre d’ion mégalithe ( des métaux) multiplier par sa valence
![]()
![]()
- Pour les oxydants et les réducteurs, x est le nombre d’électron échangé pendant réaction.
-
![]() on
aura
on
aura
![]()
6. La fraction molaire
Soit A+B une solution, avec A le soluté, B le solvant, nA le nombre de mole de A et nB le nombre de mole de B
![]()
![]()
7. Les lois de la dilution
Diluer une solution consiste à diminuer sa concentration par addition soit d’un solvant (eau) soit la même solution mais de concentration moindre.
![]()
Avec V= le volume qui est exprimé en ml et C la concentration qui est exprimé soit en Normalité, Molarité,…..
D’où par exemple ![]()
Pour un mélange des solutions, nous avons
![]() et
et ![]() notons
que pour la molarité a la place de N on met M.
notons
que pour la molarité a la place de N on met M.
Pour la préparation d’une solution nous aurons :

6.3. Loi d'action de masse
a. Définition
La loi d'action de masse (ou loi de Guldberg et Waage (1864)) est une loi qui permet de définir l'équilibre d'un système réactionnel.
Elle a été exposée en 1867 par les chimistes norvégiens Cato Guldberg et Peter Waage à partir de leur découverte commune publiée en 1864. Mais elle a surtout connu un rayonnement scientifique prometteur à partir des développements menés en 1887 par Van 't Hoff qui lui confère un rôle fondamental en chimie analytique.
Cette loi explicite les conditions de l'équilibre chimique dans la continuité des travaux de Claude Louis Berthollet, Henry Le Chatelier, Jacobus Henricus van 't Hoff et Willard Gibbs. Un système réactionnel, soumis à une réaction chimique ayant atteint un équilibre, est caractérisé par le fait que les concentrations des réactifs de départ et des produits formés sont reliées par une expression dont la valeur est constante à une température donnée.
La constante ![]() ainsi
définie est appelée constante d'équilibre de Guldberg et Waage. Ils
abordèrent également l'aspect cinétique
chimique de l'équilibre
chimique en proposant l'hypothèse que l'équilibre obtenu n'est pas
statique mais dynamique ou stationnaire : les vitesses de la réaction
directe et de la réaction inverse étant égales.
ainsi
définie est appelée constante d'équilibre de Guldberg et Waage. Ils
abordèrent également l'aspect cinétique
chimique de l'équilibre
chimique en proposant l'hypothèse que l'équilibre obtenu n'est pas
statique mais dynamique ou stationnaire : les vitesses de la réaction
directe et de la réaction inverse étant égales.
b. Loi d’action de masse relative aux équilibres homogènes
Il y a équilibre homogène lorsque le système comporte une seul phase, gazeuse ou liquide.
Soit la réaction suivante :
![]()
Selon GULDBERG et WAAGE, la vitesse d’une telle réaction est proportionnelle à la concentration des corps réagissant.
On peut exprimer les vitesses V1 et V2 de réaction directe et indirecte par les solutions suivantes :
![]()
A l’équilibre V1 = V2
d’où ![]()
Donc 
Avec : Kc : la constate d’équilibre de la réaction considéré
![]()
K1 et K2 : sont des constantes cinétiques de la réaction directe et indirecte.
Si le système est gazeux, ces concentrations sont remplacées par des pressions partielles des constituants gazeux (Pi). Rappelons que la pression partielle est égale à la pression totale multipliée par la fraction molaire du gaz considéré.
![]()
Alors l’expression de loi d’action des masses peut s’écrire :

Il existe une relation entre Kp
et Kc si on considère A,B,C et D comme de gaz parfait en utilisant la relation
suivante : ![]()
Après démonstration on aura :
![]()
Avec Δn : la variation de nombre des moles, R : la constante des gaz parfaits et T : la température absolue.
c. Loi d’action de masse relative aux équilibres hétérogènes
Pour cet équilibre on a 2 ou plusieurs phases dont une au moins est solide. Dans l’expression de la loi d’action des masses on fait intervenir que les concentrations des constituants de la phase gazeuse ou liquide.
6.4. Les équilibres Acide et Base
a) Définition
Brønsted et Lowry ont donné une définition simple des concepts d’acide et de base comme étant respectivement un donneur et un accepteur de proton. D’autres conceptions de l’acidité sont utilisées dans les milieux non protiques (milieux où l’espèce échangeable n’est pas le proton), telles la théorie de Lewis :
|
Théorie |
Acide |
Base |
Domaine d’application |
|
Arrhenius |
donneur de H+ |
donneur de HO− |
Eau |
|
Brønsted |
donneur de H+ |
accepteur de H+ |
solvant protique |
|
Lewis |
accepteur de pair e− |
donneur de pair e− |
cas général |
Exemples :
- HO− est une base d’Arrhenius, Brønsted et Lewis ;
- NH3 est une base de Brønsted et Lewis, mais pas d’Arrhenius ;
- BF3 est un acide de Lewis, mais ni d’Arrhenius, ni de Brønsted.
b) Constante d’acidité et de basicité
- Lors de la dissolution d'un acide dans l'eau entre en jeu une réaction acide-base du type : (avec AH un acide et A- sa base conjuguée)
AH + H2O = A- + H3O+
On définit alors comme constante d'acidité :
![K_{A}=\frac{\frac{[A^{-}]_{f}}{C^{0}}\cdot \frac{[H_{3}O^{+}]_{f}}{C^{0}}}{\frac{[AH]_{f}}{C^{0}}}](CHIMIE%20GENERALE%20UAC%202017_fichiers/image206.png) où C0
vaut 1 mol l-1. KA est par conséquent sans unité.
L'indice f signifie "final", c'est-à-dire à l'équilibre (on évite
d'utiliser la notation eq que les néophytes associent parfois à tort à
"équivalence").
où C0
vaut 1 mol l-1. KA est par conséquent sans unité.
L'indice f signifie "final", c'est-à-dire à l'équilibre (on évite
d'utiliser la notation eq que les néophytes associent parfois à tort à
"équivalence").
Plus la constante d'acidité est grande, plus l'acide se dissocie dans l'eau, et donc plus l'acide est fort.
Par commodité, on utilise souvent le pKa au lieu du Ka, défini ainsi :
![]()
; le pKa est souvent tabulé à 25 °C.
Ainsi, plus le pKa est petit cette fois (à ne pas confondre avec la constance d'acidité), plus l'acide est fort. Et donc plus il se dissout dans l'eau.
- Par analogie, on définit aussi la constante de basicité KB (soit B la base et BH+ l'acide conjugué) :
B + H2O = BH+ + HO- ;
On a alors :
![K_{B}=\frac{\frac{[BH^{+}]_{f}}{C^{0}}.\frac{[HO^{-}]_{f}}{C^{0}}}{\frac{[B]_{f}}{C^{0}}}](CHIMIE%20GENERALE%20UAC%202017_fichiers/image208.png)
De même
![]()
c) États de réactions entre deux couples acide/base : KA1, KA2
Lors de la réaction dans l'eau d'un acide(A1H) et d'une base (A2-) il est possible de déterminer à partir de la constante d'acidité l'état de la réaction : très peu avancée, équilibre ([A1-]=[A2-]), totale.
d) Formule générale
![]()
![]()
![]()
e) Utilisation
- Si K < 10-4, alors la réaction est très peu avancée.
- Si 10-4 < K < 104, alors il y a un état d'équilibre.
- Si K > 104, alors la réaction est totale.
f) Solubilité des sels, le produit de solubilité Ks
Le Ks mesure la solubilisation des sels dans un solvant donné. Si dans le solvant donné, le sel AB se décompose selon l'équation
![]()
alors le produit de solubilité Ks est défini par :
![]() (valeurs à saturation,
c'est-à-dire à l'équilibre entre sel précipité et sel dissous).
(valeurs à saturation,
c'est-à-dire à l'équilibre entre sel précipité et sel dissous).
Plus Ks est grand, plus le sel étudié est soluble dans le solvant.
g) Produit ionique de l’eau Ke.
1° Définition
A 25°C le pH de l’eau pure est 7. L’eau pure contient des ions oxonium H3O+ en concentration [ H3O+ ]éq = 10-7 mol.L-1, produits par la réaction :
H2O + H2O = HO-(aq) + H3O+(aq)
C’est la réaction d’autoprotolyse de l’eau qui produit autant d’ions H3O+ que d’ions HO- :
A 25°C, [ HO- ]éq = [ H3O+ ]éq = 10-7 mol.L-1
La constante d’équilibre de cette réaction Ke = [ HO- ]éq x [ H3O+ ]éq est le produit ionique de l’eau.
Elle est indépendante de la présence d’autres substances dissoutes, mais dépend de la température.
A 25°C, Ke = 10-14 donc pKe = - log Ke = 14.
Si la solution est acide [ H3O+] > [HO-], si elle est basique [HO-] > [ H3O+], mais [ H3O+].[ H3O+] = 10-14.
2° Conséquence
[ H3O+] =
10-pH et [HO-] = ![]() :
la mesure de pH permet de déterminer [ H3O+] et [HO-]
et inversement.
:
la mesure de pH permet de déterminer [ H3O+] et [HO-]
et inversement.
6.5. Notion de pH
6.5.1. Définition
Le pH ou potentiel hydrogène est une mesure de l'activité chimique des ions hydrogènes H+ (appelés aussi couramment protons) en solution. Notamment, en solution aqueuse, ces ions sont présents sous la forme de l'ion oxonium (également, et improprement, appelé ion hydronium).
Il s'agit d'une notion introduite par le chimiste danois Soerensen, permettant de caractériser l'acidité d'une solution. Par définition, le pH d'une solution est donné par la relation : pH = - log [H3O+] où [H3O+] est exprimé en mol.l-1.
L’acidité d’une solution aqueuse dépend de la concentration en ions H3O+.
En toute rigueur, pH = - log a(H3O+), mais dans les cas énumérés, l'activité de l'ion H3O+ sera assimilée à sa concentration.
Le pH d'une solution dépend de la concentration des protons dissociés, et non pas de toute la concentration en acide dans une solution.
Pour l'eau pure, les concentrations en [H3O+] et [OH-] sont égales, [H3O+] = [OH-] = 10-7 mole/l à 298 K
![]()
Dans une solution d'acide il existe un excès de protons, [H3O+] > [OH-]
![]()
A l'inverse, dans une solution de base, il existe un défaut de protons, [H3O+] < [OH-]
![]()
Ceci peut être reproduit sur une échelle de pH :

b) Domaine ou « diagramme de prédominance »
Tout couple acide/base faible est caractérisé par sa constante d’acidité :
![]()
Sachant que : pH = - log [H3O+] et pKa = - log Ka, ainsi on tire :

si pH = pKa, [Acide] = [Base]
si pH > pKa, [Acide] < [Base] : la base est l’espèce prédominante.
si pH < pKa, [Acide] > [Base] : l’acide est l’espèce prédominante.
On appelle "diagramme de prédominance" une représentation graphique délimitant les domaines de concentration.
Le diagramme de prédominance correspondant à l’exemple précédent est le suivant :

4.Détermination du pH
a) pH d’un monoacide fort.
Lorsque l’on dissout dans l’eau un acide fort HA (HNO3, H2SO4, HBr, HCl, HI..) de concentration initiale Co, on a :

Dans le cas de solutions peu diluées (Co > 3.10-7 mol.l-1) le milieu est suffisamment acide pour que [OH-] issu de l’autoprotolyse de l’eau soit négligeable devant [H3O+].
On a: [H3O+] = Co soit: pH = -log [H3O+] = -log Co
La formule : pH = - log Co est valable jusqu’à un pH égal à 6,5
Exemple : Acide chlorhydrique HCl de concentration initiale : Co = 10-3 mol.l-1
pH = - log Co = - log 10-3 = 3
b) pH d’une monobase forte.

Dans le cas de solutions peu diluées (Co > 3.10-7 mol.l-1) le milieu est suffisamment basique pour que [H3O+] issu de l’autoprotolyse de l’eau soit négligeable devant [OH-].

Cette formule est valable pour un pH calculé supérieur à 7,5 : pH > 7,5
Exemple : solution de soude NaOH de concentration initiale : Co = 10-3 mol.l-1
pH = 14 + log Co = 14 + log 10-3 = 11
c) pH d’un monoacide faible.
Un acide faible (CH3COOH, HF, acides organiques..) est partiellement dissocié dans l'eau.Un acide faible HA a un pKa compris entre 0 et 14 ΔG°ass > 0). Soit une solution d’un monoacide faible HA de concentration initiale Co. Deux équilibres se produisent simultanément :
![]()
Quatre espèces sont présentes à l’équilibre, en concentration : [HA], [A-], [H3O+]et [OH- ]. On peut écrire quatre relations entre ces quatre inconnues :

Le calcul conduit à une une équation du 3ème degré en [H3O+] :
[H3O+] 3 + Ka [H3O+] 2 - (Ke + KaCo) [H3O+] - KeKa = 0
D’où la nécessité de simplifier en faisant simultanément les deux approximations suivantes :
- si l'acide est assez concentré (> 10-6 M) pour négliger l'autodissociation de l'eau. L’équation (4) devient alors : [H3O+] = [A-]
- si l'acide est suffisamment faible donc peu dissocié. Ainsi, [A-] << [HA].

Exemple : Calculer le pH d’une solution d’acide acétique CH3COOH (pKa = 4,8) de concentration Co = 10-1 mol.l-1 : pH = 1/2 (4,8 + 1) = 2,9
d) pH d’une monobase faible.
Une base faible B a un pKb compris entre 0 et 14 (ΔG°diss > 0).
Soit une solution d’une monobase faible B telle que l’ammoniaque NH3(aq) de concentration initiale Co. Deux équilibres se produisent simultanément :
![]()
Quatre espèces présentes sont à l’équilibre, en concentration : [BH+], [B], [H3O+] et [OH-].

Le calcul conduit à une équation du 3e degré en [H3O+] :
[H3O+]3 + (Co + Ka) [H3O+]2 - Ke [H3O+] - Ke Ka = 0
D’où la nécessité de simplifier en faisant simultanément les deux approximations suivantes :
- si la base est assez concentrée (> 10-6 M) pour négliger l'autodissociation de l'eau.Ainsi, [H3O+] << [OH-]. L’équation (4) devient alors : [BH+] = [OH-]
- si la
base est suffisamment faible pour être peu protonée (pKb
![]() 2). On
peut envisager deux cas selon que ces conditions sont réunies ou non. La
protonation de la base est faible; elle fixe peu les protons de l’eau; la
concentration de l’acide conjugué BH+ est négligeable devant celle
de la base B : [BH+] << [B].
2). On
peut envisager deux cas selon que ces conditions sont réunies ou non. La
protonation de la base est faible; elle fixe peu les protons de l’eau; la
concentration de l’acide conjugué BH+ est négligeable devant celle
de la base B : [BH+] << [B].
L’équation (3) devient alors : Co = [B]. En remplaçant [B] et [BH+] dans Ka, on a :
![]()

Exemple : Calculer le pH d’une solution d’ammoniaque NH3 (pKa = 9,2) de concentration Co = 10-2 mol.l-1 : pH = 1/2 (14 + 9,2) = 10,6
e) pH d’une solution d’ampholyte.
Considérons une solution d’un sel NaHA (par exemple NaHCO3) de concentration C. La dissociation totale du sel dans l’eau s’écrit :
![]()
HA- est un ampholyte puisqu’il est l’acide du couple HA-/A2- et la base du couple H2A/HA-. Deux réactions faisant intervenir HA- se produisent :


f) pH des solutions salines.
pH d’une solution d’un sel d’acide fort et de base forte.
Exemple : NaCl (chlorure de sodium) . HCl + NaOH → NaCl + H2O
On a donc un sel d’acide fort et de base forte. En solution aqueuse, il y a dissolution totale du sel :
![]()
Cl-: base conjuguée (très faible) d’un acide fort (HCl). Cl-est un ion indifférent ou spectateur; il ne participe à aucun équilibre acido-basique. Na+ : ion spectateur, acide conjugué (très faible) d’une base forte (NaOH); il ne participe à aucun équilibre acido-basique.
![]()
pH d’une solution d’un sel d’acide fort et de base faible.
Exemple : NH4Cl (chlorure d’ammonium). HCl + NH3→ NH4Cl
On a donc un sel d’acide fort et de base faible
En solution aqueuse, il y a dissolution totale du sel :
![]()
Cl- : ion spectateur, base conjuguée très faible d’un acide fort (HCl) et NH4+ : acide conjugué (faible) de la base faible NH3 (pKa = 9,2)
Þ pH (NH4Cl) = pH (NH4+ ) Þ pH d' un acide faible

pH d’une solution d’un sel d’acide faible et de base forte.
Exemple : CH3COONa (acétate de sodium). CH3COOH + NaOH → CH3COONa + H2O
On a donc un sel d’acide faible et de base forte. En solution aqueuse, il y a dissolution totale du sel :
![]()
Na+ : ion spectateur, acide conjugué très faible d’une base forte (NaOH) et CH3COO- : base conjuguée (faible) de l’acide faible CH3COOH (pKa = 4,8)
Þ pH(CH3COONa) = pH(CH3COO- )Þ pH d' une base faible

pH d’une solution d’un sel d’acide faible et de base faible.
Exemple: CH3COONH4 (acétate d’ammonium) de concentration C (C = [CH3COONH4])
CH3COOH + NH3 → CH3COONH4. On a donc un sel d’acide faible et de base faible.En solution aqueuse, il y a dissolution totale du sel :
![]()

Or, le mélange d’un acide faible et d’une base faible donne une solution faiblement acide ou faiblement basique Þ pH est voisin de 7.
On montre que si : [H3O+] << C et [OH-] << C, en faisant le produit Ka1.Ka2 :
![]()
g) Solution tampon.
Le pH du sang humain doit être fixé dans un domaine relativement étroit autour de pH = 7,4 (7,35-7,45) afin d'assurer la survie cellulaire. Il peut être maintenu dans ces limites grâce à des systèmes tampon faisant intervenir des carbonates, des phosphates ou des protéines plasmatiques par exemple.
Le pouvoir tampon du sang, indépendamment de son rôle dans le métabolisme va intervenir par exemple pour minimiser les conséquences d'une ingestion accidentelle d'acide.
Une solution tampon est une solution capable d'absorber une certaine quantité d'acide ou de base sans entraîner une forte variation de pH. Une solution tampon est un mélange dans des proportions égales ou voisines d'acide faible AH et de sa base conjuguée A- (acide faible en solution) ou de base faible et de son acide conjugué (base faible en solution). Il existe une large gamme de solutions tampon.

Exemple: HA + sel NaA (solide)


En général le pH d’une solution tampon est compris entre 4 < pH (solution tampon) < 10. Pour des raisons d’efficacité tampon : les concentrations C1 de l’acide faible AH et C2 du sel NaA sont relativement élevées Þ > 10-2 mol.l-1 donc > à [H3O+] et [OH-].
Etant donné que [H3O+] et [OH-] sont négligeables devant [AH] et [NaA], on peut écrire :
C1 = [AH] où AH : acide faible, peu dissocié dans l’eau
C2 = [A-] où NaA : sel entièrement dissous dans l’eau
![]()
Un couple acide-base conjuguée ne permet pas de réaliser des solutions tampons de n’importe quelle valeur du pH. L’effet tampon est en effet maximum pour pH = pKa.
Propriétés des solutions tampons.
Les solutions tampons ont la propriété de minimiser les variations de pH provoquées par :
· une addition (modérée) d’acide ou de base.
· une addition (modérée) de solvant (eau): dilution.
Exemple : Ajoutons 0.1 mol de HCl à 1 litre d’eau pure. Le pH initial est de 7.00. Après addition du HCl, le pH est de 1. Le pH varie de 6 unités.
Ajoutons maintenant 0.1 mol de HCl à une solution tampon contenant 1 M en acide acétique et 1 M en acétate de sodium. Le pH initial de la solution est
pHinitial = pKa + log ([base]/[ acide]) = 4.75 – log (1/1) = 4.75. Le HCl ajouté réagit avec la base CH3COO– et redonne de l’acide acétique non dissocié: [base] = 1 – 0.1 = 0.9, [acide] = 1 + 0.1 = 1.1. Le nouveau pH sera : pH = pKa + log (0.9 / 1.1) = 4.75 – 0.09 = 4.66. Le pH n’aura varié que de 0.09 unité.
Nous comparons ici des ajouts d'acide ou de base à une solution tampon avec les mêmes ajouts à de l'eau pure.
Soit un litre d'une solution tampon 1 M en acide acétique, CH3CO2H et 0,3 M en acétate de sodium, CH3CO2Na.
Après ajout de 100 ml de HCl 1 M :
![]()
L'équilibre est complètement déplacé vers la droite, donc :
[CH3CO2H] = (1 + 0,1) / 1,1 = 1,0 M et [CH3CO2-] = (0,3 - 0,1) / 1,1 = 0,18 M.
pH = 4,76 + log (0,18 / 1) = 4,02.
Après ajout de 100 ml de NaOH 1 M :
![]()
L'équilibre est complètement déplacé vers la droite, donc
[CH3CO2H]
= (1 - 0,1) / 1,1 = 0,82 M et [CH3CO2-] = (0,3
+ 0,1) / 1,1 = 0,36 M.
pH = 4,76 + log (0,36 / 0,82) = 4,40.
Pour préparer une solution tampon acide, il faut choisir un acide dont le pKa ne diffère pas de plus d'une unité par rapport à la valeur de pH désirée.
Exemple : Préparation d'une solution tampon de pH = 5,2. Le pKa (CH3CO2H / CH3CO2-) = 4,76, le mélange acide acétique (CH3CO2H) / acétate de sodium (CH3CO2-) peut être utilisé.
Selon la relation d'Henderson-Hasselbalch, pH = pKa + log ([A-] / [AH])
5, 2 = 4, 76 + log ([A-] / [AH]), 0, 44 = log ([A-] / [AH]), le rapport [A-] / [AH] = 10-0, 44 = 2, 75 ainsi le pH = 4, 76 + log (16,7·10-2 / 5·10-2) = 5,28. Pour préparer une solution tampon de pH = 5,2 pour un volume donné, il faut utiliser 2,75 moles d'acétate de sodium pour une mole d'acide acétique.
Les solutions tampons sont utilisées pour étalonner les pH-mètres, pour contrôler le pH de solutions où ont lieu des réactions chimiques. De même, dans le milieu vivant, de nombreuses réactions enzymatiques ne peuvent avoir lieu que dans un domaine de pH très restreint. Comme certaines d'elles consomment ou au contraire produisent des ions hydronium H3O+, il est nécessaire que des systèmes tampons interviennent pour réguler le pH.
En solution aqueuse, les équilibres sont les suivants pour H3PO4:

Le principal tampon intracellulaire et dans le milieu rénal est le tampon phosphate. Il exerce un effet tampon dans les milieux intracellulaires et est présent dans l'urine.
A pH sanguin (7,2), c'est le deuxième couple acido-basique H2PO4-/HPO42- qui joue le rôle de tampon. Sa capacité tampon est maximale lorsque le pH est proche du pKa2 du couple H2PO4-/HPO42-.

A ce pH=7,2, les autres espèces H3PO4 et PO43- sont négligeables.
Pour une solution 0,15 M de tampon phosphate à pH = 7,2 :
![]()
Si une réaction enzymatique consomme 0,01 M d'ions H3O+, le système tampon compense cette perte (équilibre déplacé vers la droite) :
![]()
À la fin de la réaction :

D’où une faible variation de pH. En l'absence d'un système tampon le pH serait passé de 7,2 à 12; or, à un pH aussi basique, l'activité enzymatique est impossible.
On entend par tampon protéique l'ensemble des groupements protéiques qui se comportent comme des ampholytes. Le rôle des protéines est assez réduit dans le plasma. Au pH plasmatique, les fonctions acides carboxyliques des protéines sont sous forme carboxylate qui assurent l'effet tampon. Il faut tenir compte du pKa moyen des fonctions acide carboxyliques/carboxylates.
L'hémoglobine Hb comporte plusieurs sites pouvant jouer un rôle de tampon dans le sang, on considère donc un pKa moyen, celui-ci variant en fonction de la forme oxygénée ou non de l'Hb:

L'ensemble des différents systèmes tampons évoqués ci-dessus lutte contre les variations de pH au sein de l'organisme en se tamponnant mutuellement : c'est le principe d'isohydrie
6.5.Applications
a) Mélange d'acides forts
Exemple : quel est le pH d'une solution obtenue en mélangeant 250 ml de HClO4 0,4 M et 350 ml de HCl 0,1 M ?
Reponse : nt = (0,25·0,4) + (0,35·0,1) = 0,135 mol; [H3O+]t = 0,135 / (0,25 + 0,35) = 0,225 M et ainsi le pH = -log [H3O+]t = -log (0,225) = 0,648
b) Mélange d'un acide fort et d'acide faible
Exemple : quel est le pH d'une solution obtenue en mélangeant 500 ml de CH3CO2H 0,03 M et 200 ml de HCl 0,04 M ?
nt = (0,2·0,04) = 0,008 mol, [H3O+]t = 0,008 / (0,2 + 0,5) = 1,14.10-2 M et ainsi le pH = - log [H3O+]t = -log (1,14·10-2) = 1,94
c) Mélange d'acides faibles
Exemple : quel est le pH d'une solution obtenue en mélangeant 100 ml de CH3CO2H (0,015 M, pKa = 4,7) et 200 ml de ClCH3CO2H (0,03 M, pKa = 2,9).
C1t = (C CH3CO2H. VCH3CO2H) / (VCH3CO2H + VCLCH3CO2H) = (0,015·0,1) / (0,1 +0,2) = 5·10-3 M
C2t = (0, 2·0,03) / (0,1 + 0,2) = 2·10-2 M
[H3O+] = (10-4,7·0,005 + 10-2,9·0,02)½ = 5·10-3 M ainsi le pH = -log [H3O+] = -log (0,005) = 2,3
d) Mélange de bases fortes
Exemple : quel est le pH d'une solution obtenue en mélangeant 150 ml de NH2- 0,2 M et100 ml de NaOH 0,3 M ?
nt = (0,2·0,15) + (0,3·0,1) = 6·10-2 mol, [OH-]t = 0,06 / (0,15 + 0,1) = 0,24 M
pOH = -log [OH-] = 0,62 et pH = 14 - pOH = 13,38
e) Mélange d'une base forte et une base faible
Exemple : quel est le pH d'une solution obtenue en mélangeant 500 ml de NH3 0,03 M et 200 ml de NaOH 0,04 M ?
nt = (0,2·0,04) = 0,008 mol [OH-]t = 0,008 / (0,2 + 0,5) = 1,14·10-2 M
pOH = -log [OH-]t = 1.94 et pH = 14 - pOH = 12,06
f) Mélange de bases faibles
Exemple : quel est le pH d'une solution obtenue en mélangeant 100 ml de CH3CO2- (0,015 M, pKb = 9,24) et 200 ml de NH3 (0,03 M, pKb = 4,76)
C1t (CH3CO2-) = (0,015·0,1) / (0,1 + 0,2) = 5·10-3 M
C 2t (NH3) = (0,2·0,03) / (0,1 + 0,2) = 2·10-2 M
[OH-] = (10-9,24 · 0,005 + 10-4,76 · 0,02)½ = 6·10-4 M , pOH = -log [OH-] = 3,22 et donc le pH = 14 - pOH = 10,78
6.6.Problèmes
1. Quelle est la variation du pH d’une solution de HCl 0.1 M qui est diluée 10 fois avec
de l’eau? Réponse: Le pH varie de 1 à 2.
2. Quel est le pH d’une solution tampon CH3COOH (acide acétique)/ CH3COONa (acétate de sodium), chacuns de deux composants ayant une concentration de 0.2 M? (le pKa de l’acide acétique est 4.77). Réponse: pH = 4.77
3. Soient les solutions qui suivent (à 25°C) :
a) Quel est le pH d’une solution de NaOH 0.1 M ?
b) Quel est le pH d’une solution qui contient 5 g de NaOH pour 2 litres d’eau ?
c) Quel est le pH d’une solution NaOH 10-13 M ? Cette réponse vous surprend-elle?
d) Quel le pH d’une solution de HCl 0.001 M ?
e) Quel est le pH d’une solution de HNO3 1.10-14 M. Le résultat trouvé vous surprendil?
Réponses:
a) pH = 14 - pOH à 25°C. pOH = -log[OH-] = - log(0.1) ⇒ pOH = 1 et pH =13.
b) [NaOH] = 5 g.l-1 ⇒ [NaOH] = 5/MM (NaOH) = 0.125 mol dans 2 L ⇒ [OH-] = 0.0625 M et pOH = - log (0.0625) = 1.204 ⇒ pH = 12.796.
c) [NaOH] << [OH-] dû à la dissociation de l’eau (= 10-7 M à 25°C) ⇒ pOH = pH ≈
d) pH = 3
e) pH ≈ 7 [les H+ fournis par la dissociation de l’eau sont les plus abondants ([HNO3] << 10-7 M)].
4. Quel est le pH d’une solution de NH3 0.1 M? (la constante de basicité Kb de NH3 vaut 1.71.10-5). Réponse: pH = 11.12
5. On veut obtenir une solution de pH = 3.0 à partir d’un mélange de vinaigre et d’eau pure. Quel est le mélange à réaliser (mL de chaque liquide) pour obtenir 1 litre de cette solution, sachant que le vinaigre contient 5% en masse d’acide acétique (CH3COOH, masse spécifique = 1 kg/L) ? Le pKa de CH3COOH est égal à
Réponse:Le pH d’un acide faible vaut : pH = ½(pKa -log[AcOH]0). Donc [AcOH]0 = 0.0589 mol⋅l-1.
-AcOH à 5 % contient 50 g d’acide acétique pur pour 1 litre d’eau ⇒ 50/MM(AcOH) = 50/60 = 0.833 mole d’AcOH dans 1 litre d’eau.
-Pour calculer le nombre de ml d’AcOH à 5 % nécessaire pour préparer 1 litre d’AcOH à 0.0575 M, on applique la règle de 3 : 0.0575 = (x ⋅ 0.833)/1000 ⇒ x ≈ 71 mL et par conséquent 1000-71 mL d’eau seront nécessaires.
6.6. Electrolyse
C’est la décomposition d’une substance par le passage de courant électrique. La solution est appelée électrolyte.
L'électrolyse est une méthode qui permet de réaliser des réactions chimiques grâce à une activation électrique. C'est le processus de conversion de l'énergie électrique en énergie chimique. Elle permet par ailleurs, dans l'industrie chimique, la séparation d'éléments ou la synthèse de composés chimiques. La première électrolyse par courant continu (électrolyse de l'eau) a été réalisée le 2 mai 1800 par deux chimistes britanniques, William Nicholson (1753-1815) et Sir Anthony Carlisle (1768-1842), quelques jours après l'invention de la première pile électrique (publication soumise le 20 mars 1800 dans une lettre en français au président de la Royal Society, Joseph Banks) par Alessandro Volta et grâce à celle-ci. Par ailleurs, onze ans auparavant J. R. Deiman et A. Paets van Troostwijk avaient déjà réalisé une électrolyse de l'eau au moyen d'une machine électrostatique et d'une bouteille de Leyde sans réussir à interpréter la réaction observée.
L'électrolyse est utilisée dans divers procédés industriels, tels que la production de dihydrogène par électrolyse de l'eau, la production d'aluminium ou de chlore, ou encore pour le placage d'objets par galvanoplastie.
A. Principe
La matière à décomposer ou à transférer est dissoute dans un solvant approprié, ou fondue de sorte que ses ions constitutifs soient disponibles dans la solution.
- Une différence de potentiel électrique est appliquée entre deux électrodes immergées dans cette solution.
- La cathode est le siège d'une réduction et, l'anode le siège d'une oxydation. Le potentiel de l'anode étant supérieur -ou égal dans une pile court circuitée- au potentiel de la cathode on peut dire que l'anode est la borne positive et que la cathode est la borne négative. Notons que ces bornes sont inversées dans le cas d'une pile.
- Lors du passage d'un courant électrique continu, les électrodes attirent à elles les ions de charge opposée. Mais il est faux de dire que l'électrolyse se résume à l'oxydation des anions à l'anode et à la réduction des cations à la cathode. En effet, il est aussi possible d'oxyder des cations à l'anode.

Schéma de la synthèse d'hydrogène et d'oxygène par électrolyse de l'eau.

Représentation symbolique d'un électrolyseur dans un circuit
B. Lois de Faraday (électrochimie)
Les lois sur l'électrolyse de Faraday sont basées sur des recherches que Michael Faraday publie en 1834.
Énoncé des lois
Première loi : La quantité de substance libérée lors de l’électrolyse à une électrode est proportionnelle au temps et au courant électrique (ce qui équivaut à la charge).
Seconde loi : Les poids de divers corps séparés aux électrodes par la même quantité d'électricité sont entre eux comme leurs équivalents chimiques.
Formule mathématiques
Les lois de Faraday peuvent être représentées par la forme suivante :
![]()
avec :
- m la masse de la substance libérée à l'électrode en grammes
- Q la charge électrique totale passée à travers la substance
- F = 96485 C mol−1 ≈ 96.500C mol-1, la constante de Faraday
- M la masse molaire de la substance
- z la valence de la substance
M/z correspond à l'équivalent de la substance libérée.
Pour la première loi de Faraday M, F et z sont des constantes dont m augmente en fonction de Q.
Pour la seconde loi de Faraday Q, F et z sont des constantes donc m varie en fonction de M/z (l'équivalent).
Dans le
cas simple d'une électrolyse à courant constant, ![]() donc
donc
![]()
ce qui donne
![]()
avec
- n la quantité de matière libérée : n = m/M
- t le temps durant lequel le courant est appliqué
Dans
les cas plus compliqués où le courant varie, la charge électrique Q est
l'intensité électrique I(![]() ) intégrée en fonction du temps
) intégrée en fonction du temps ![]() :
:
![]()
avec t le temps d'électrolyse.
d. L’équation de NERNST
En se basant sur les considérations thermodynamique, Nernst (chimiste et physicien allemand : 1864-1941) a donné l’équation qui permet de calculer le potentiel d’électrode en fonction de la température et des concentrations.
Soit un système redox dont la demi-équation redox est la suivante :

Avec : [ox] : concentration de la forme oxydée
[red] : concentration de la forme réduite
R : Constante des gaz parfait= 8,31 J.mol-1 T-1
F : Faraday= 95500coulomb
T : température en degré absolue
n : nombre d’électrons échangé entre la forme oxydée et la forme réduite.
A 25°C : T=25+273=298K
Lnx=a→x=ea
Logx=logea=a loge


Selon Nernst :


Par convention :
ü E0 : potentiel redox normal du système=constante pour un couple redox donné
ü E0 : positif pour les couples dont la forme oxydée est meilleur oxydant que H+ et négatif pour les couples dont la forme réduite est meilleur réducteur que H2.
ü Une forme oxydée ou réductrice qui s’échappe du milieu réactionnel par précipitation ou volatilisation ne figure pas dans l’équation de Nernst.
ü La concentration du solvant n’intervient pas dans la formule de Nernst.
MEDIAGRAPHIE
1. PANNETIER G. (1969). Chimie physique générale. Atomistique, liaisons chimiques et structures moléculaires. Ed. Masson. p. 309.
2. PANNETIER G. (1969). Chimie physique générale. Atomistique, liaisons chimiques et structures moléculaires. Ed. Masson. p. 310.
3. S. ZUMDAHL, S.( 1998) : Chimie générale, 2e éd., Les Éditions CEC Inc.,p. 243-249
4. ALAIN GERSCHEL (2012) : Liaisons intermoléculaires : Les forces en jeu dans la matière condensée, Paris, EDP Sciences, 2e éd., 288 p. (ISBN 9782759802784, lire en ligne [archive]), p. 12
5. P. W. ATKINS(1992) : Chimie Générale, Inter Editions
6. K. BADIBANGA (2009): Cours Approfondie de Chimie Générale, inédit UCG/Bbo/Fac Agro.
7. Paul ARNAUD(1990) : Cours de chimie physique, Dunod
8. P.W. ATKINS(1985) : Physical Chemistry, 3e éd., Oxford University Press



